Downloaded 11 times


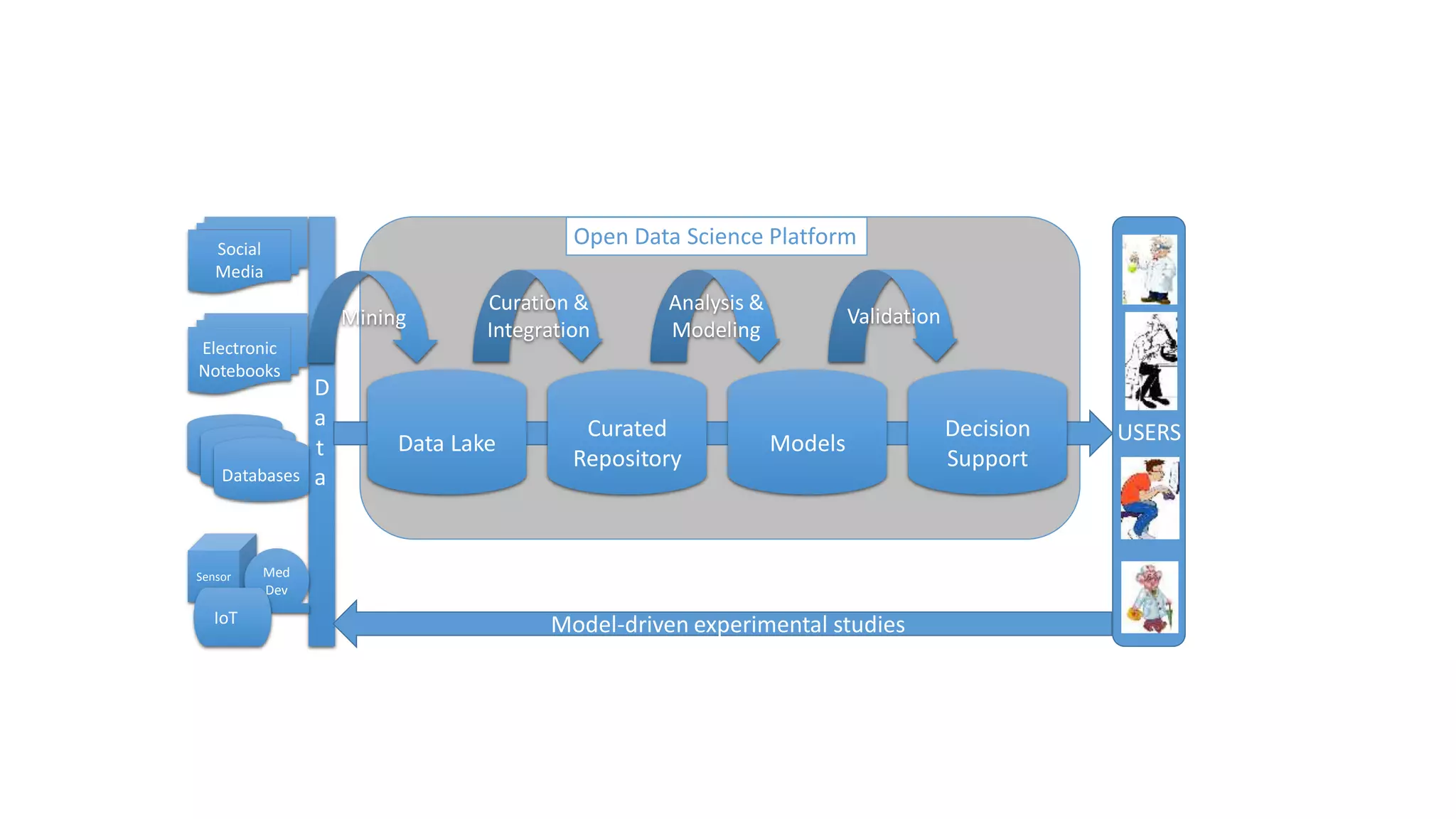
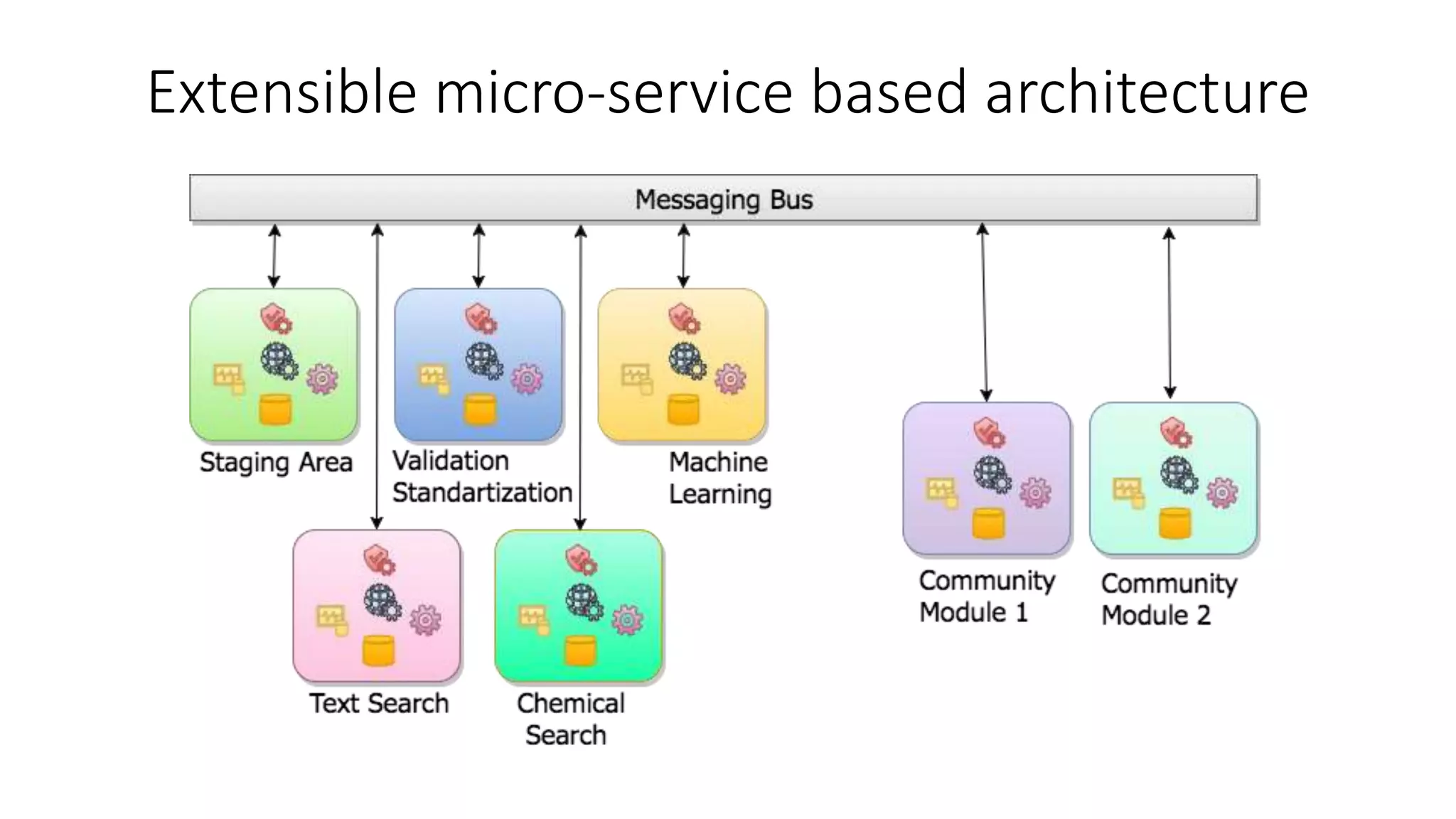
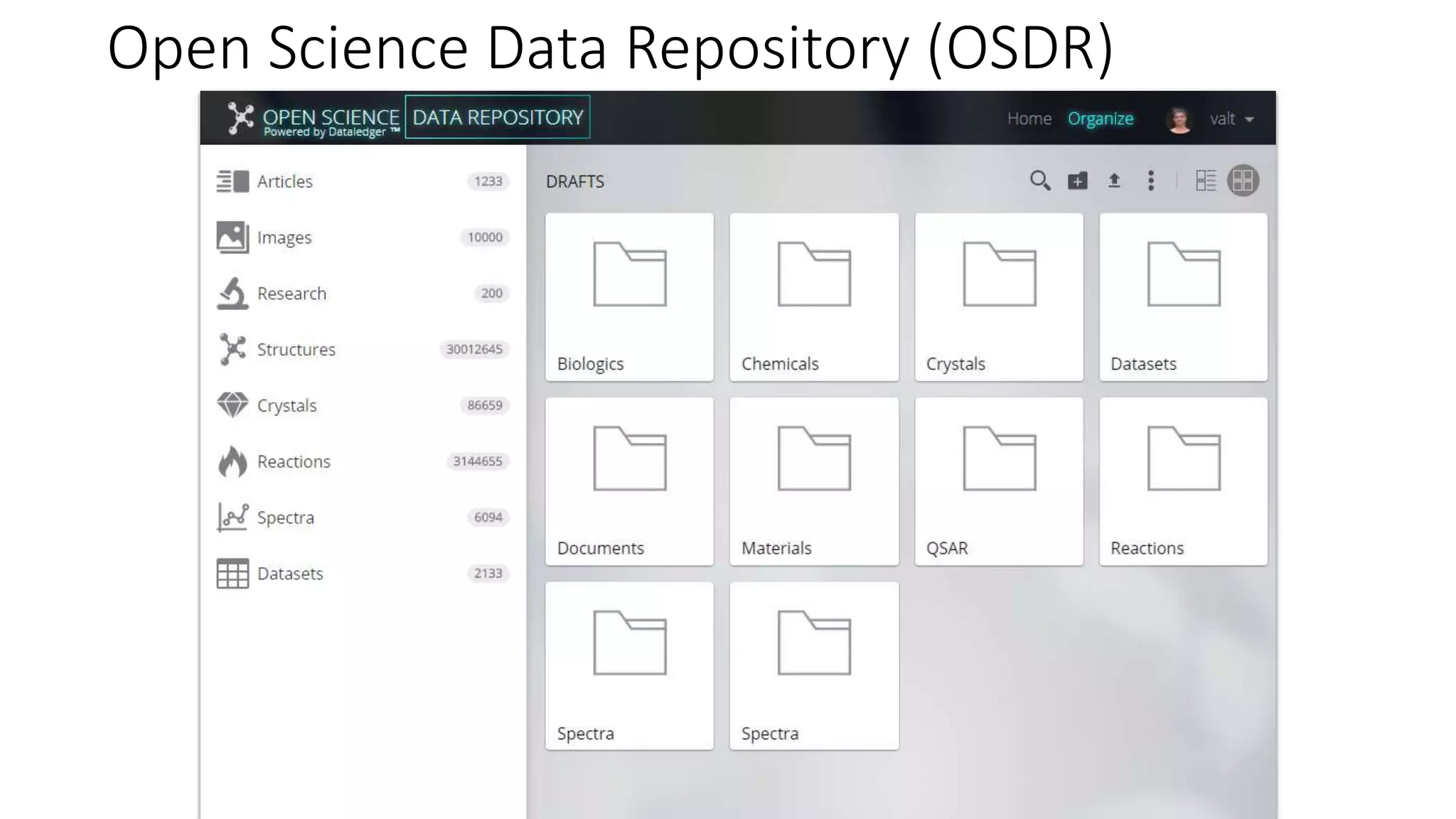
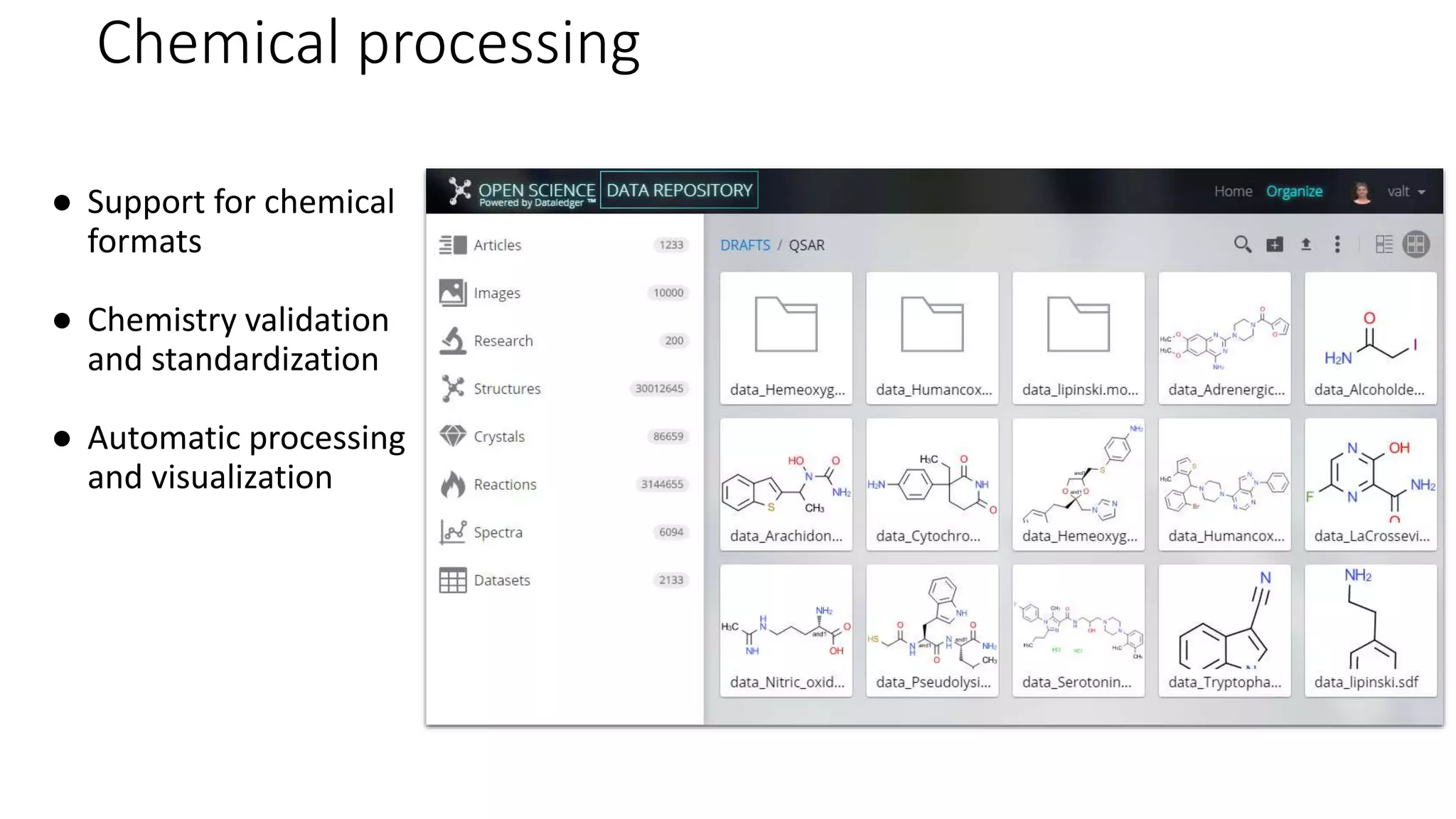
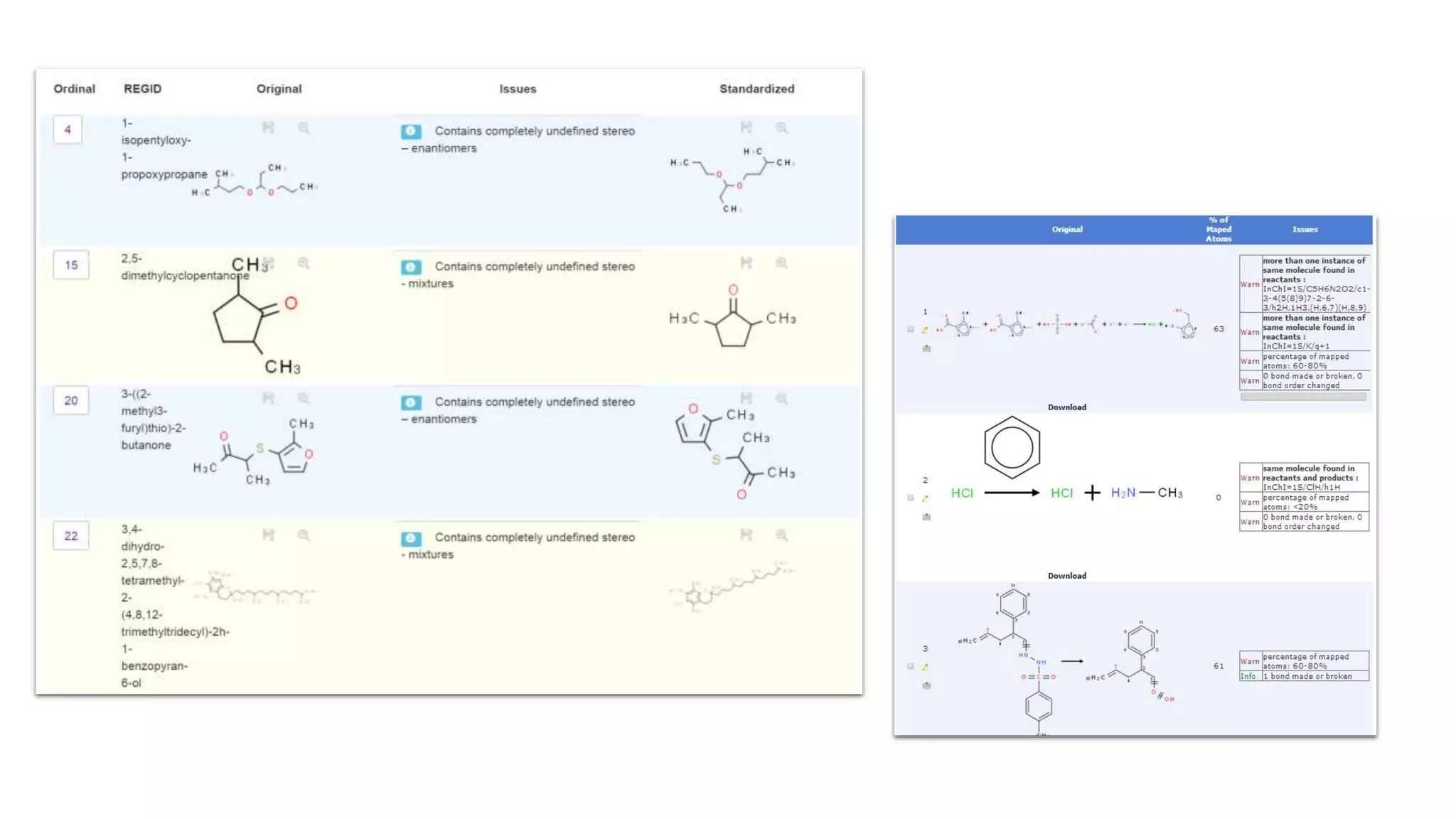
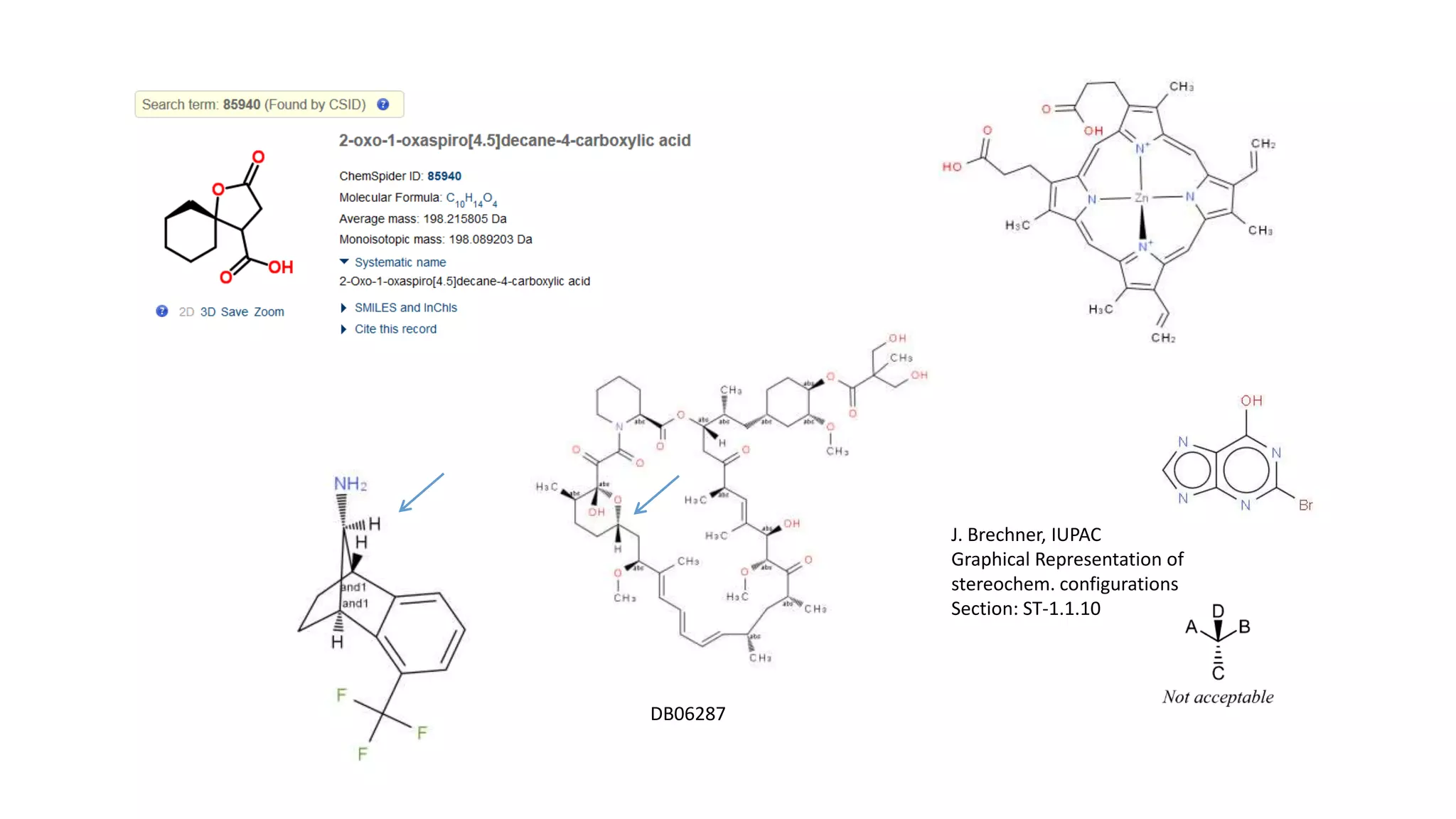
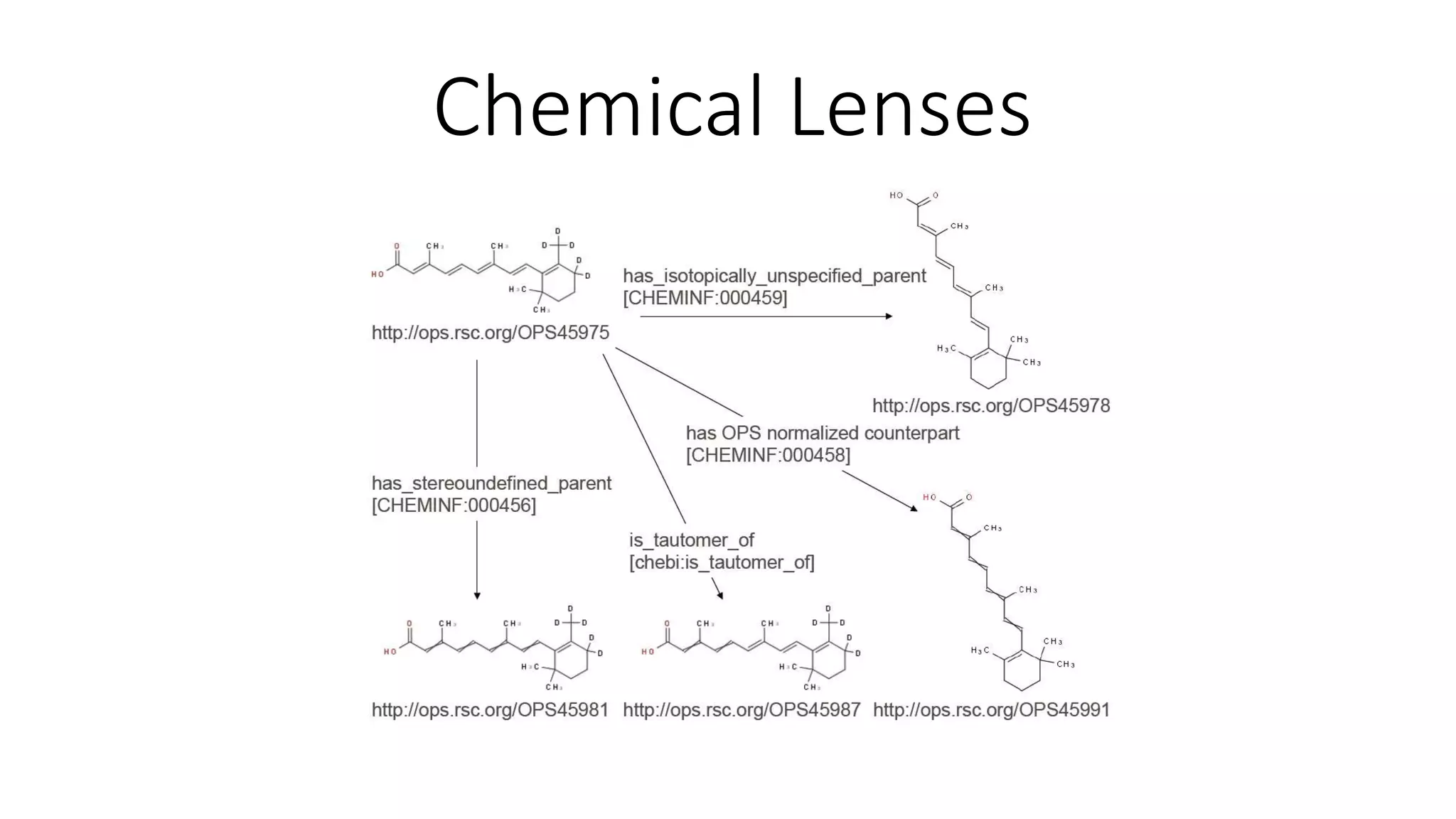
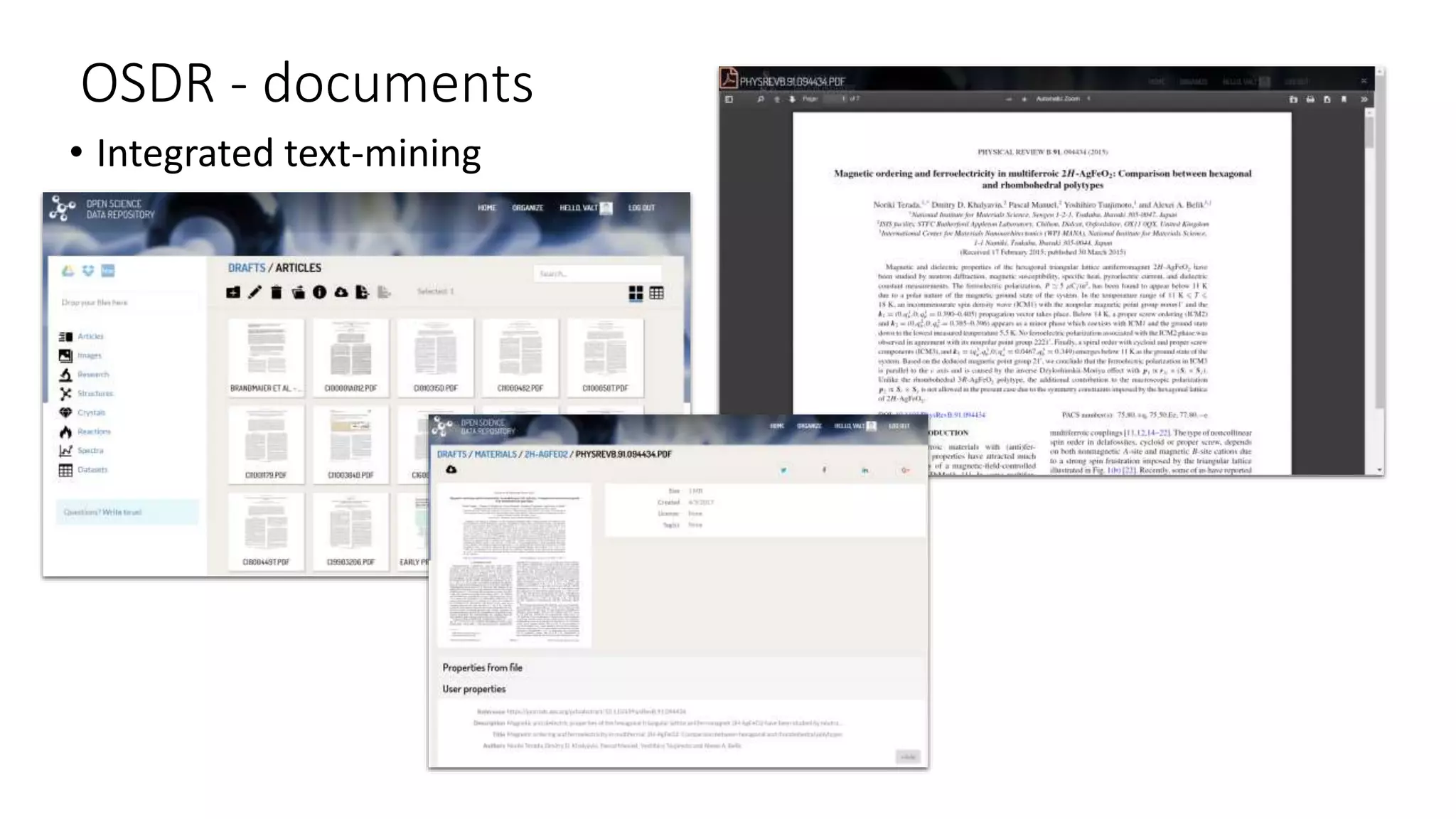

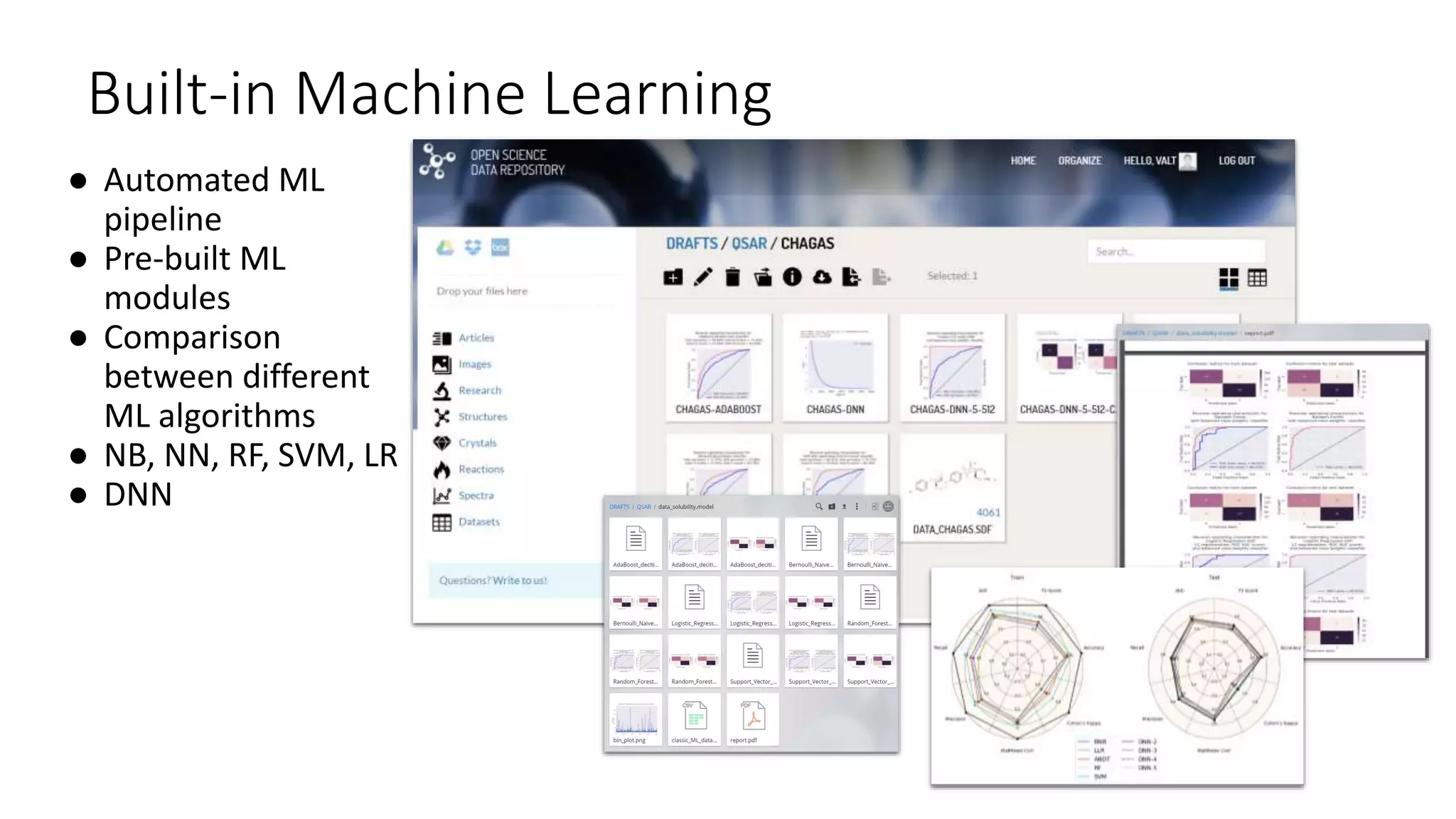
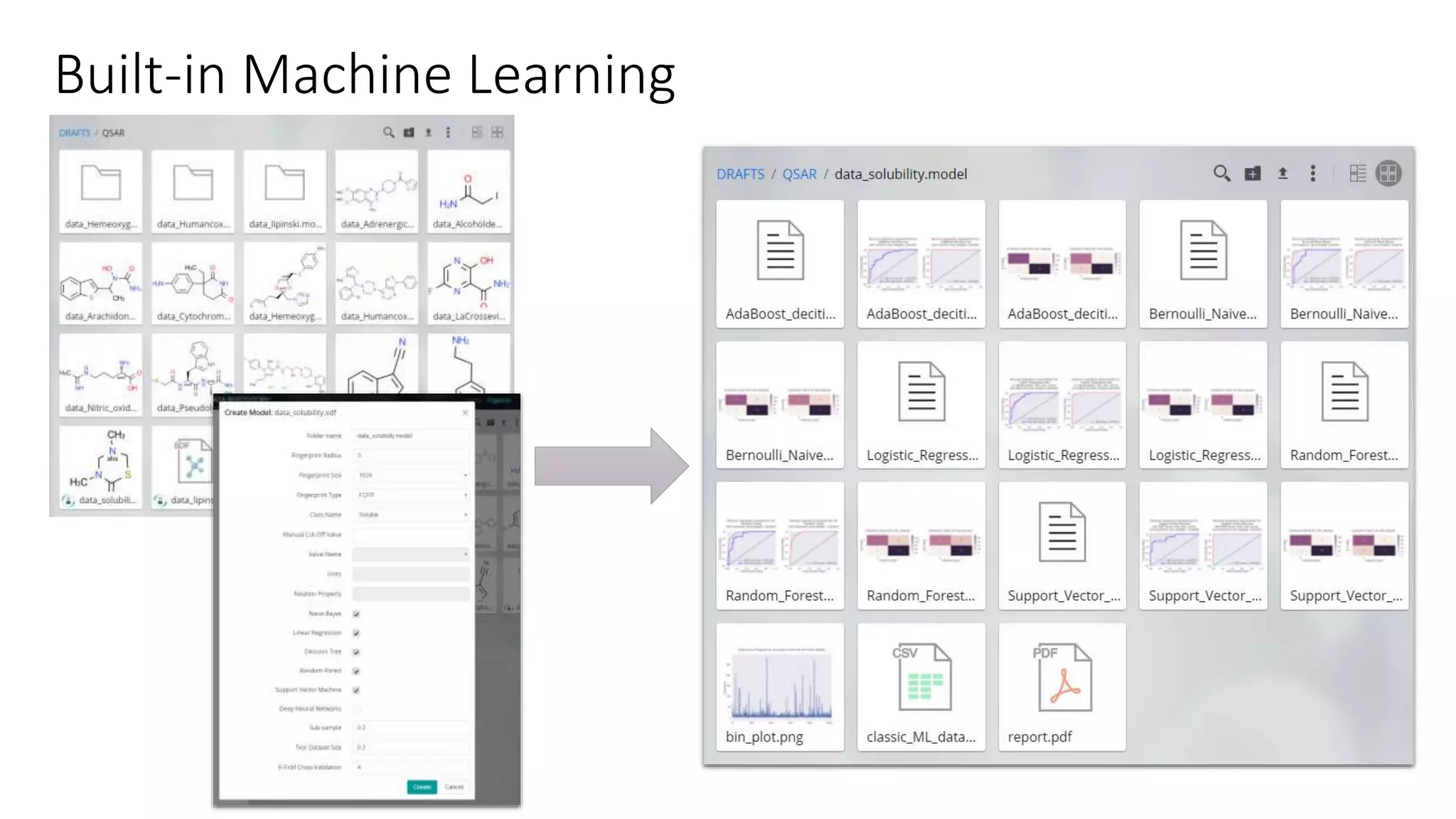

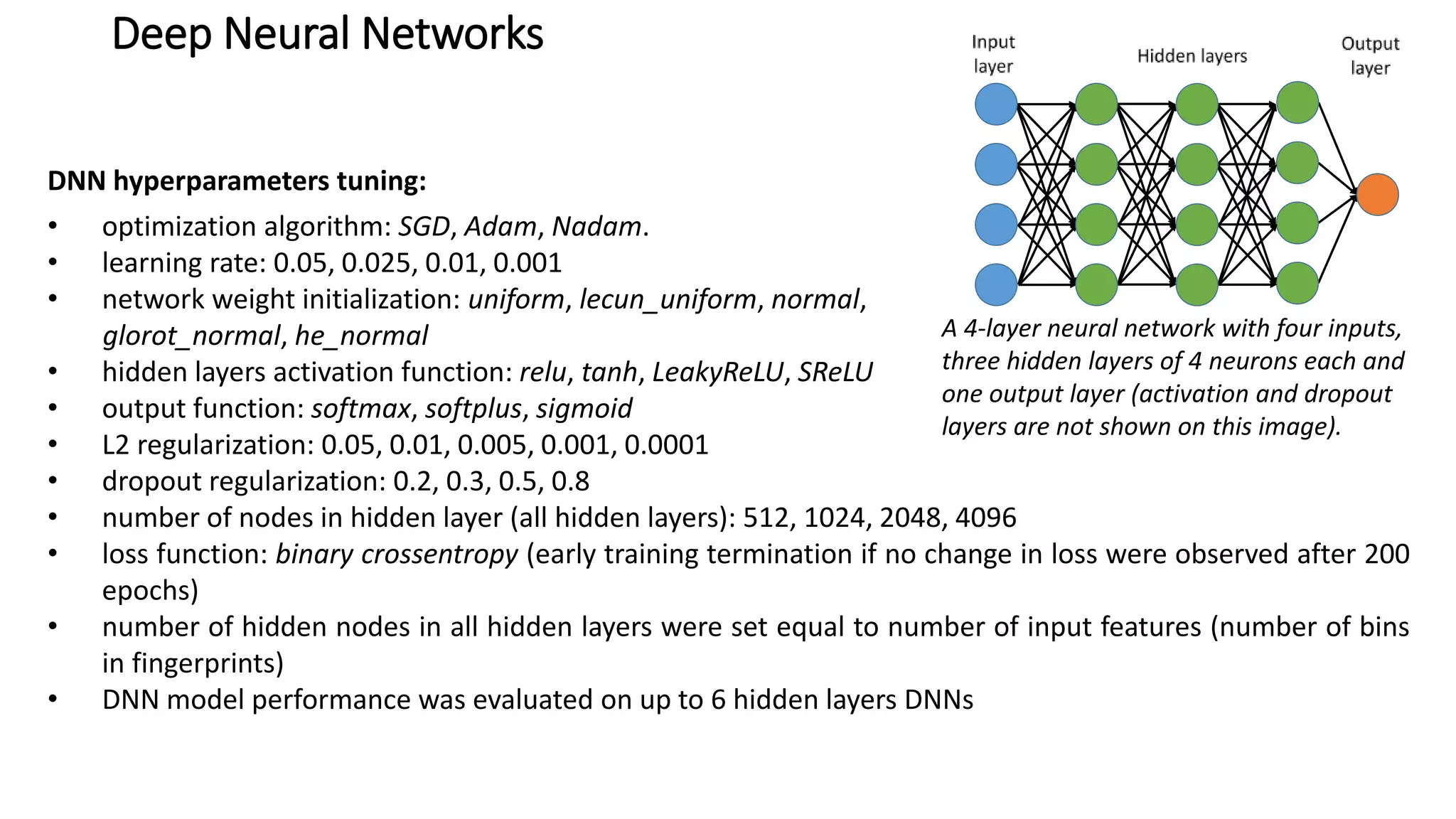

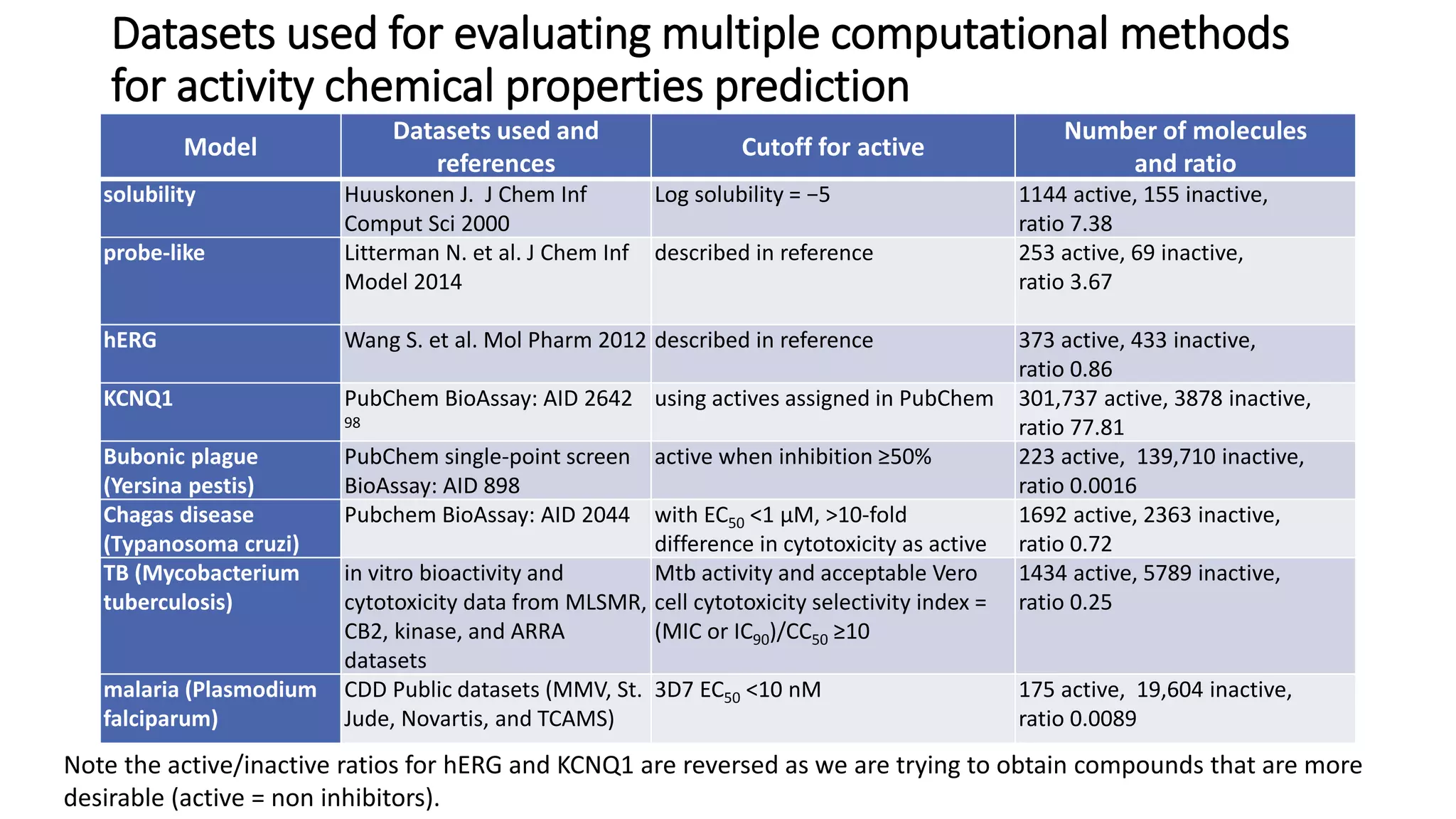
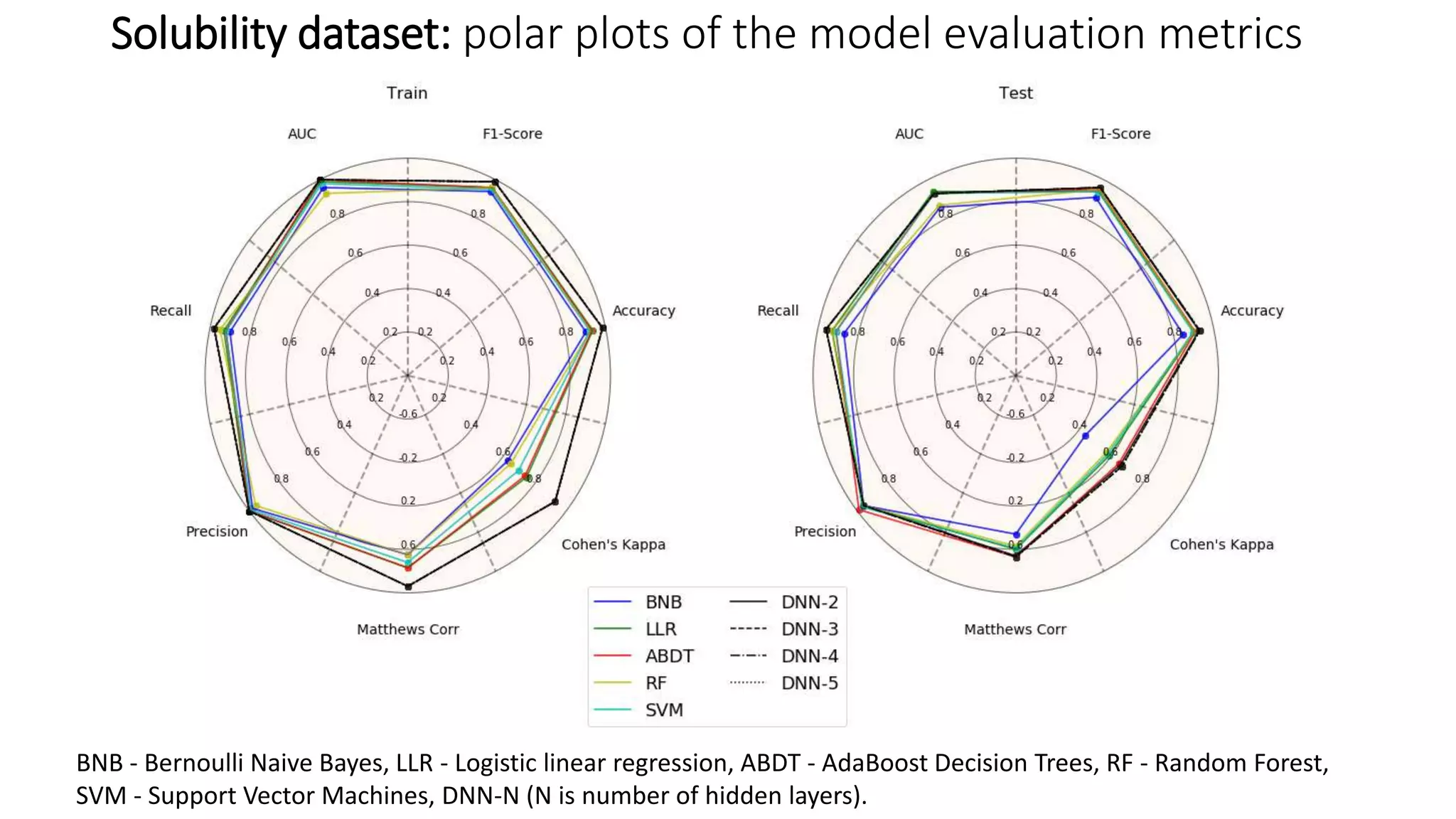
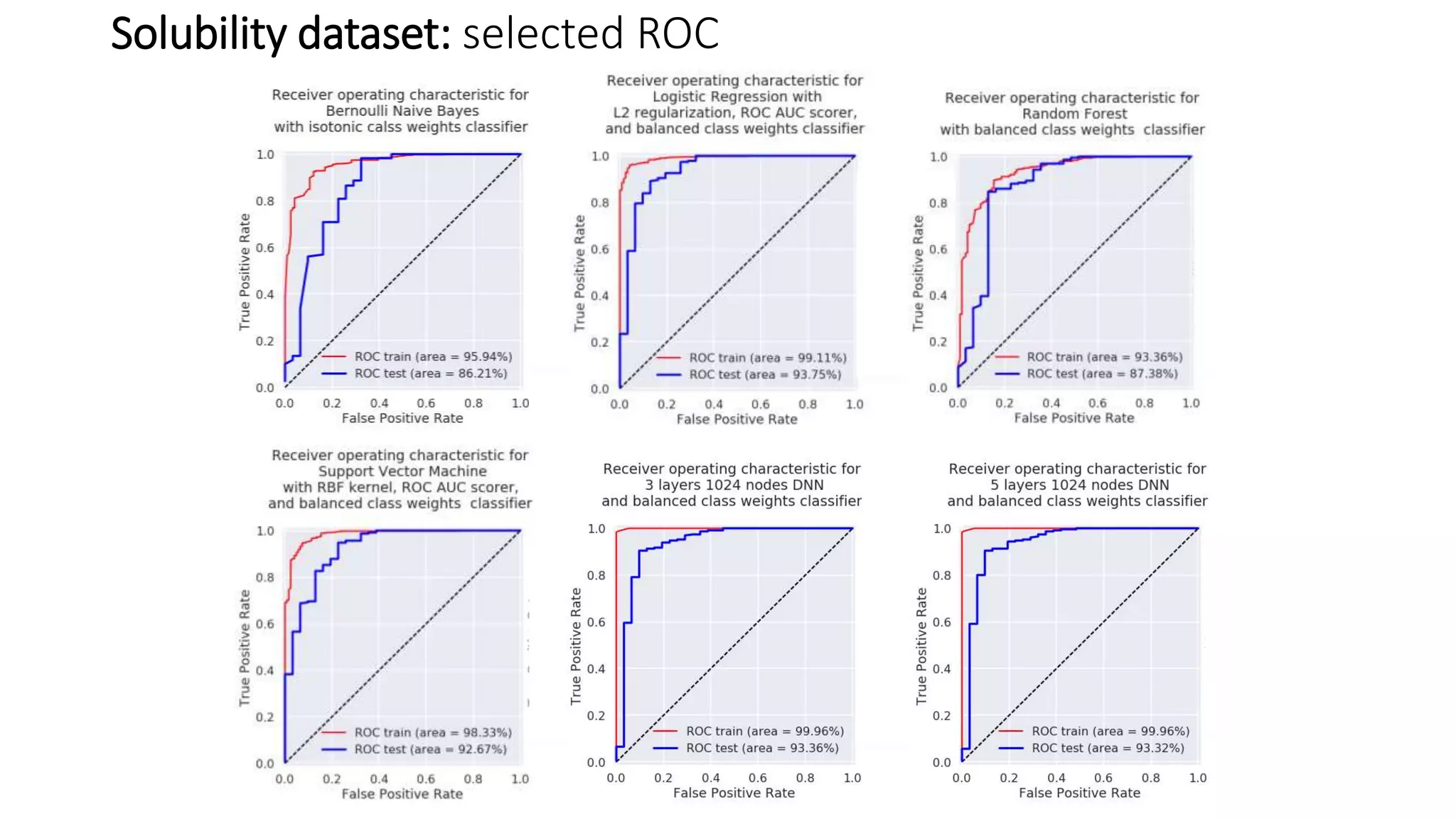
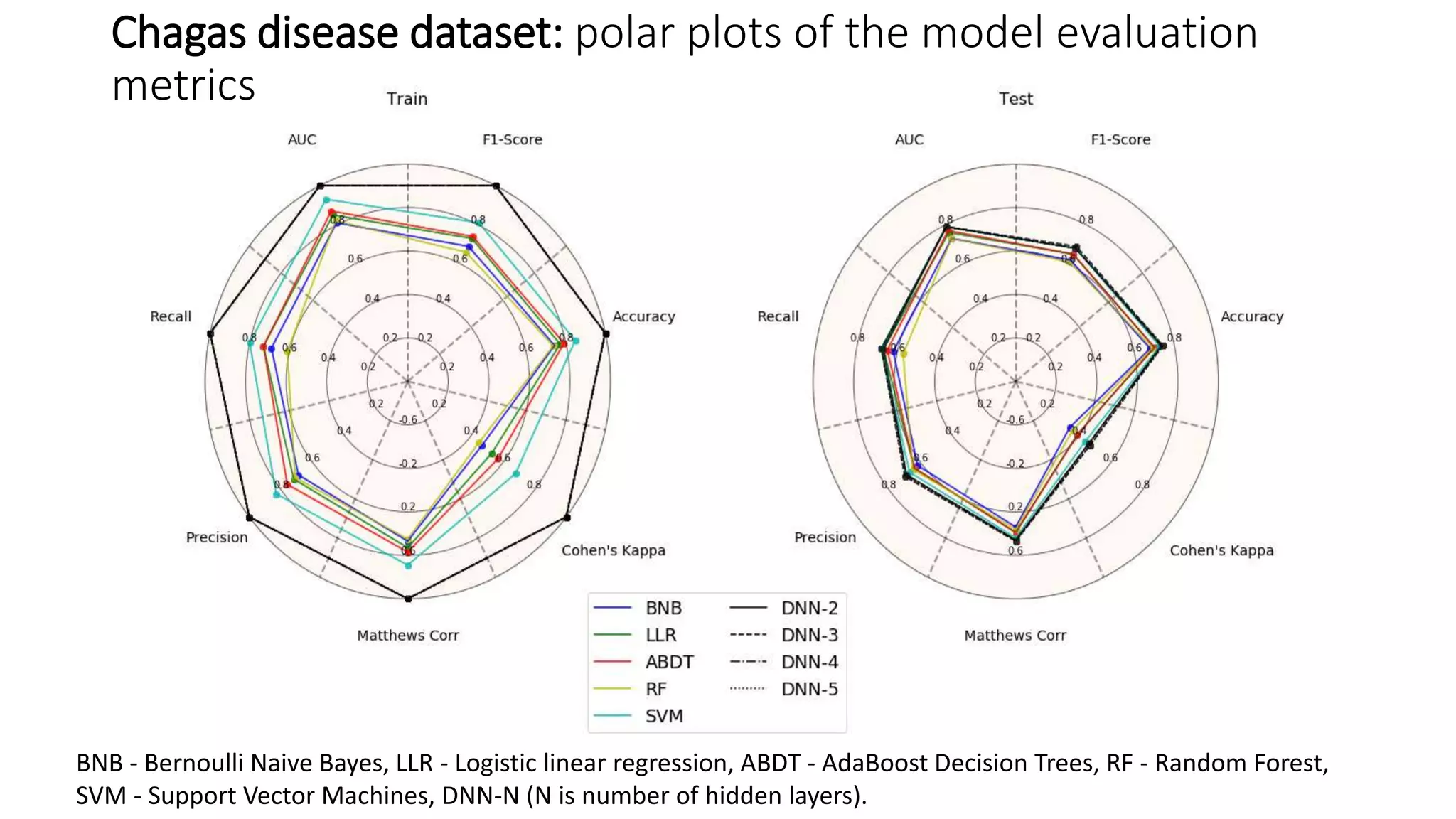
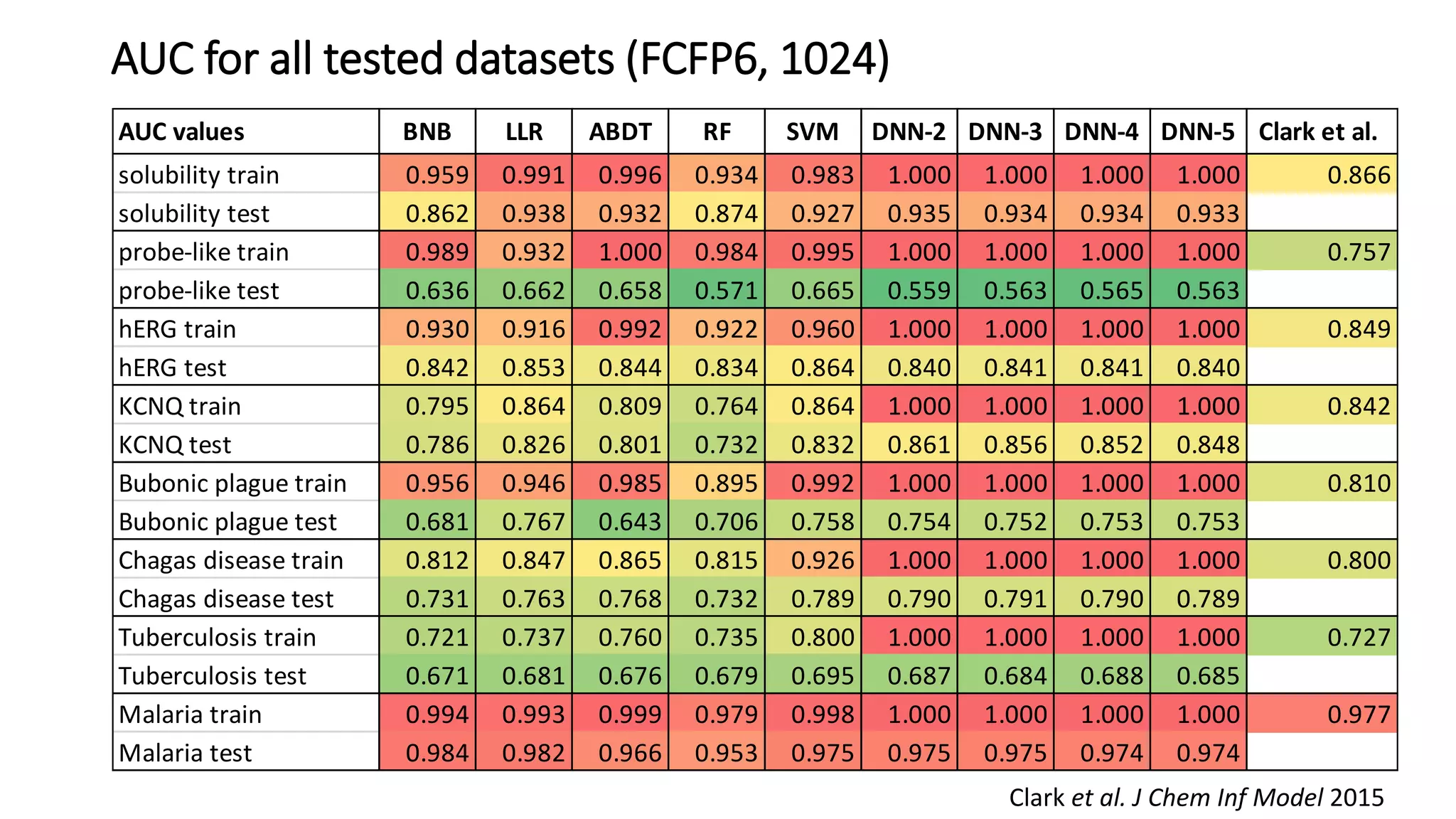
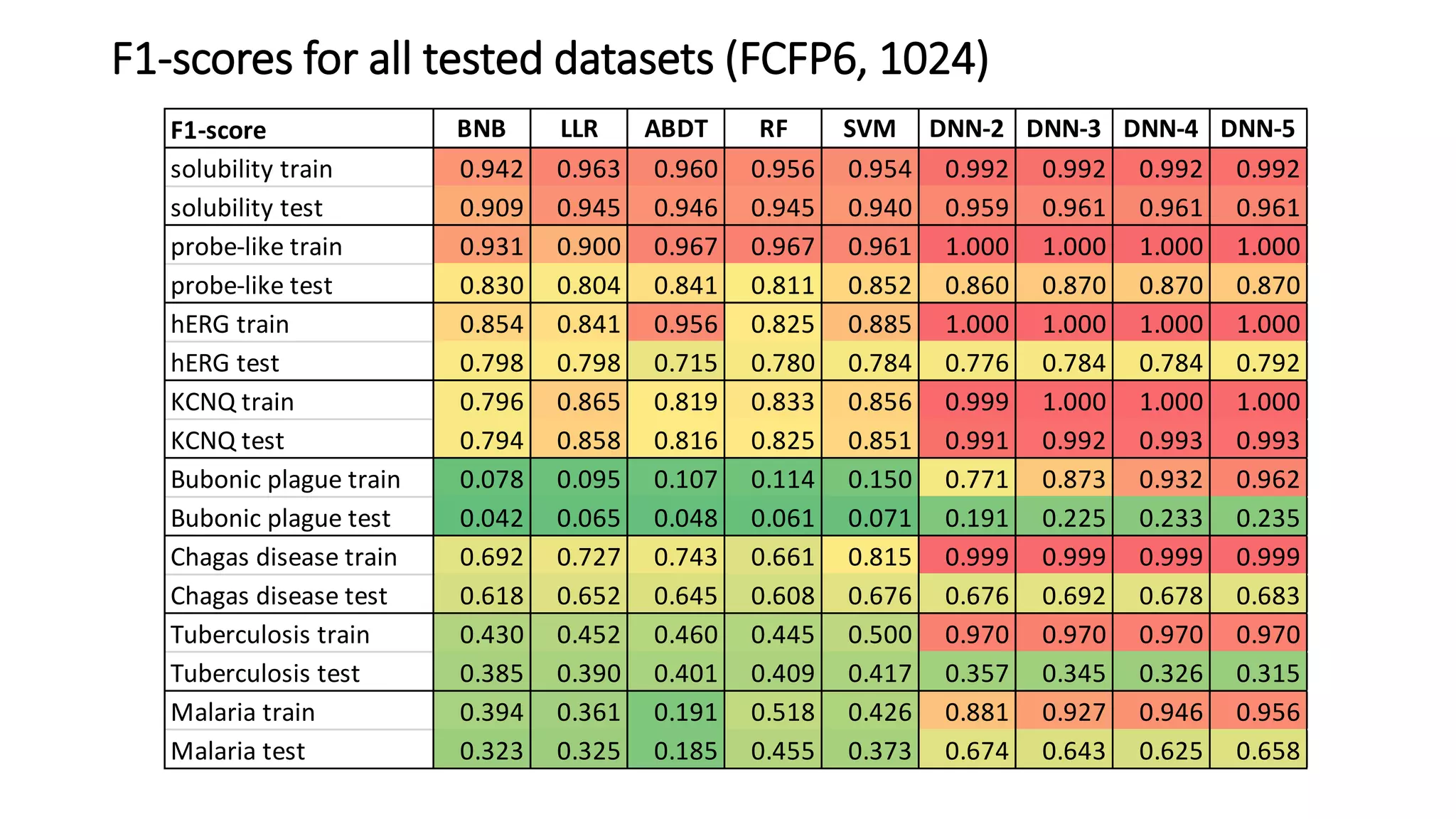
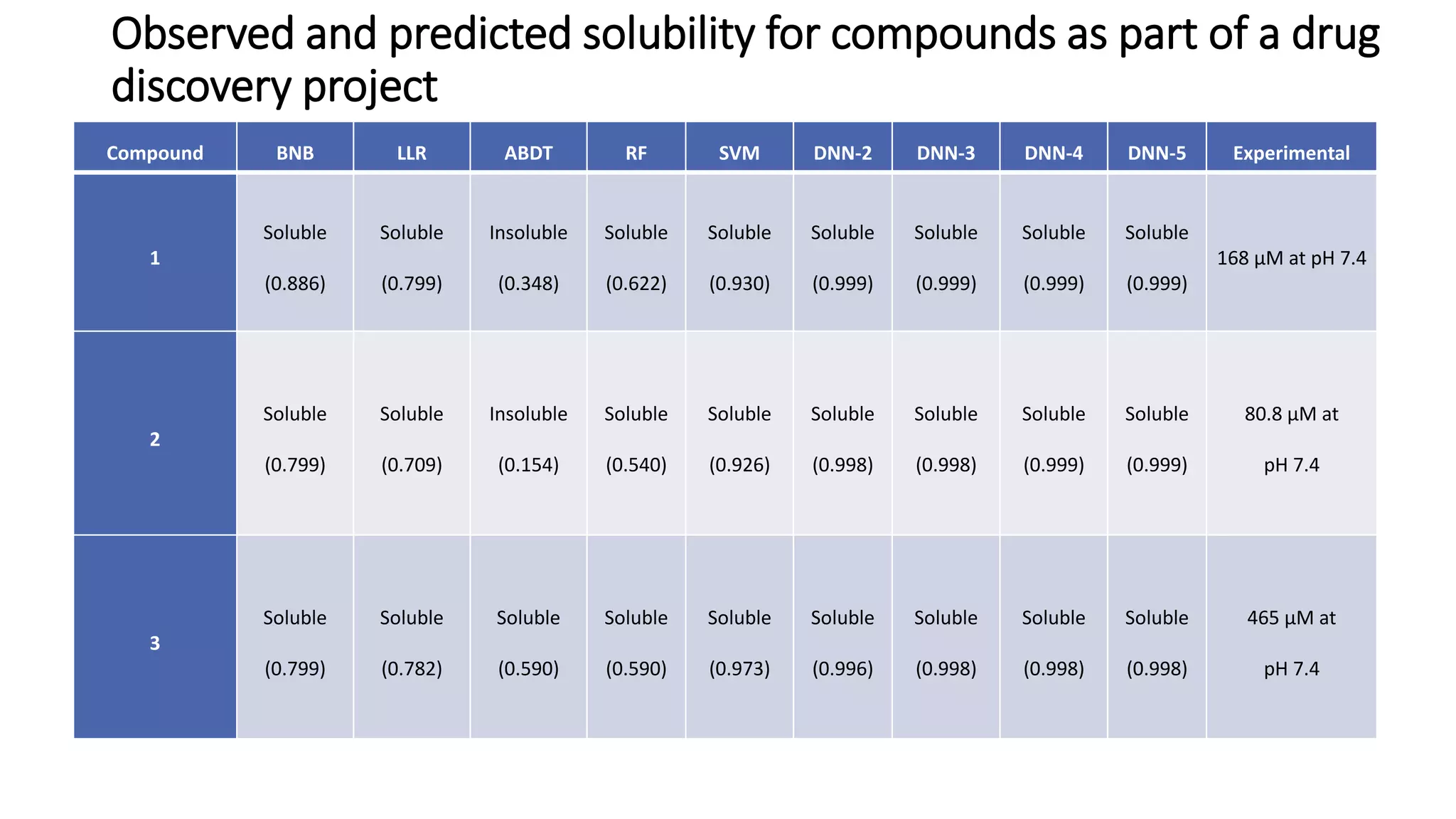






























The document describes the development and evaluation of a machine learning toolkit for an open science data repository, implementing both classic machine learning methods and deep neural networks (DNNs) for chemical properties prediction. The performance of various models, including metrics like ROC, AUC, and F1 scores, indicates that DNNs generally outperform classic models in real-world drug discovery tasks such as assessing solubility. Future research is suggested to explore additional models and descriptors to enhance predictive accuracy.



































































