Downloaded 51 times









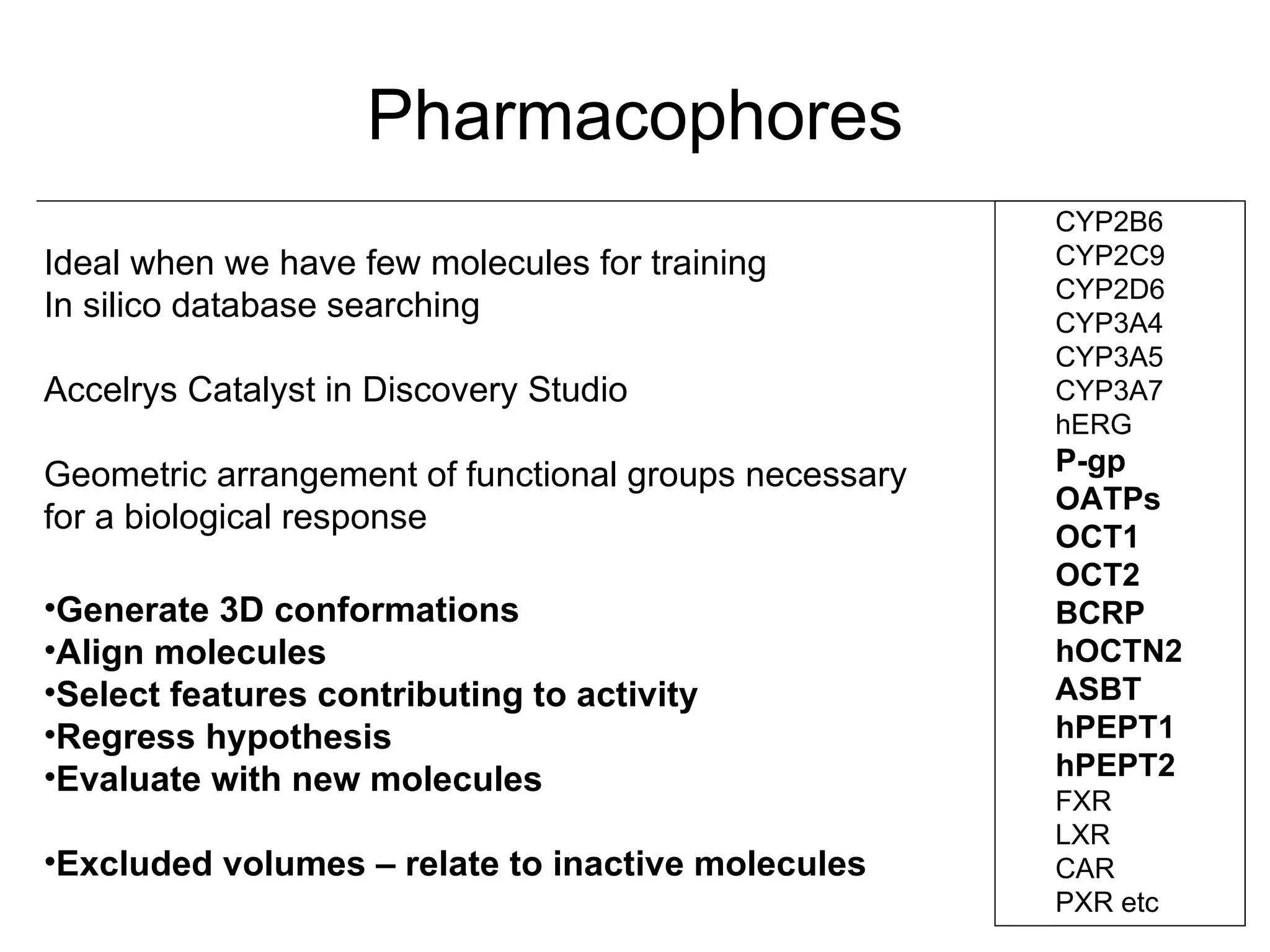
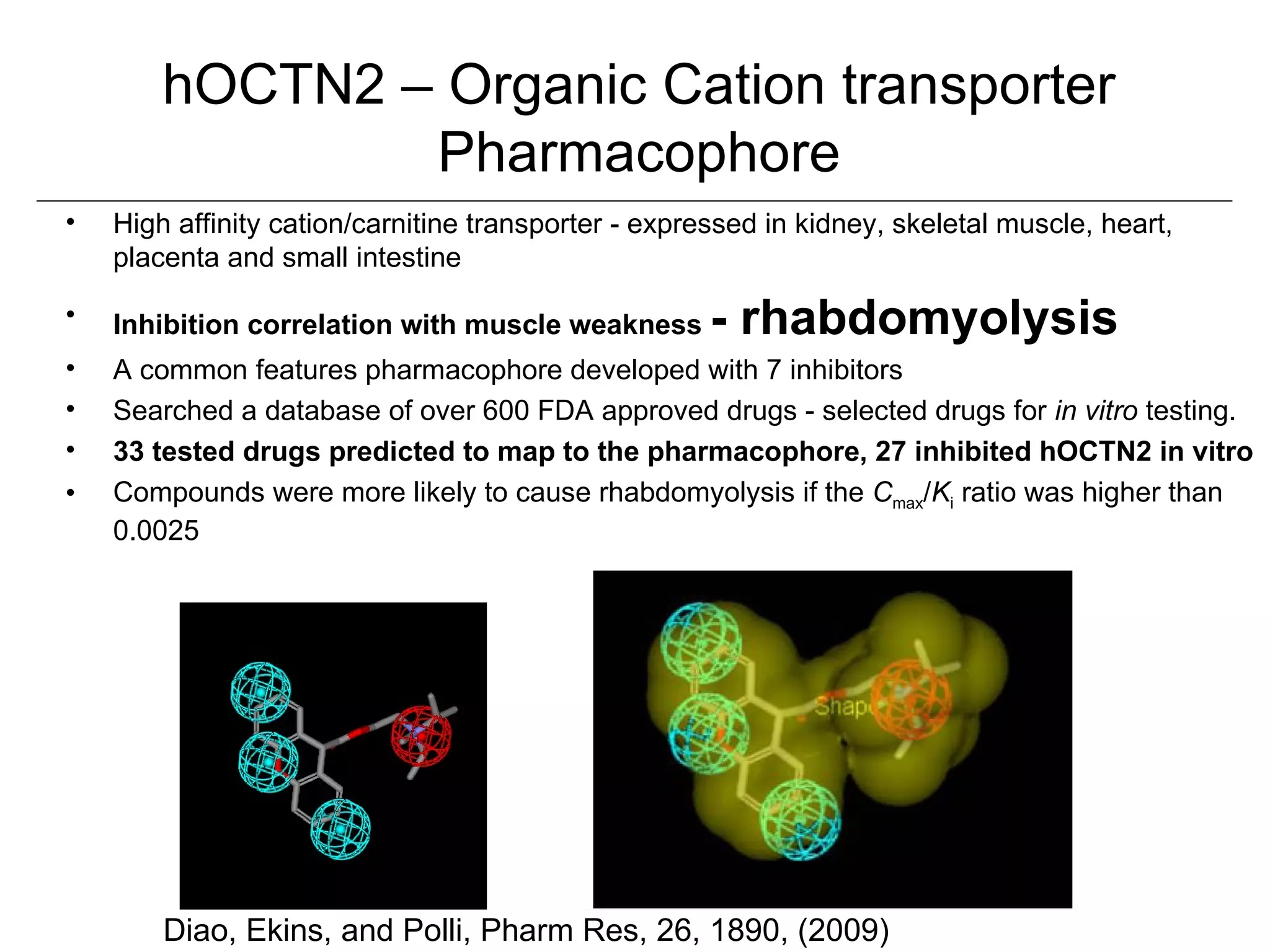
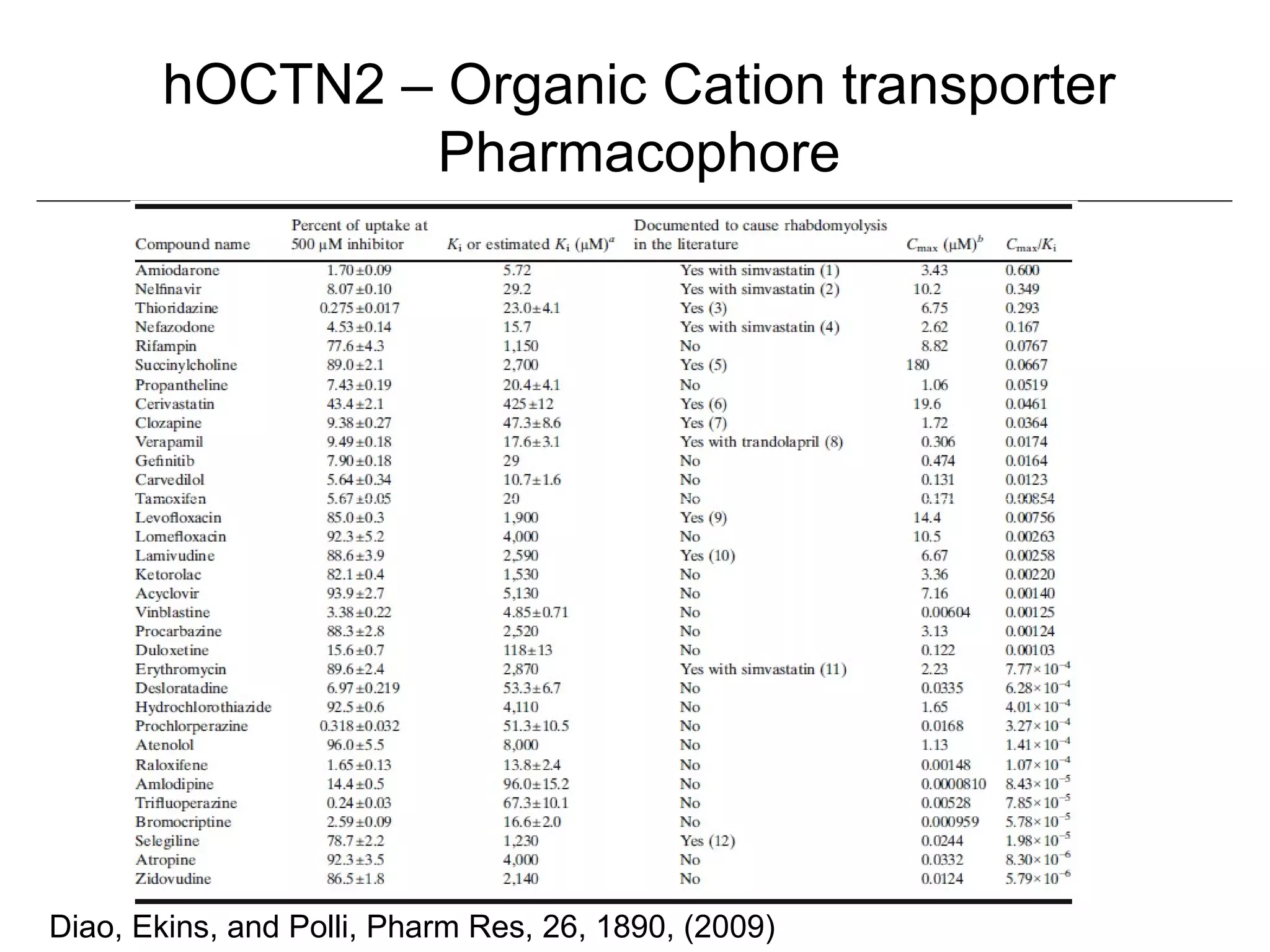





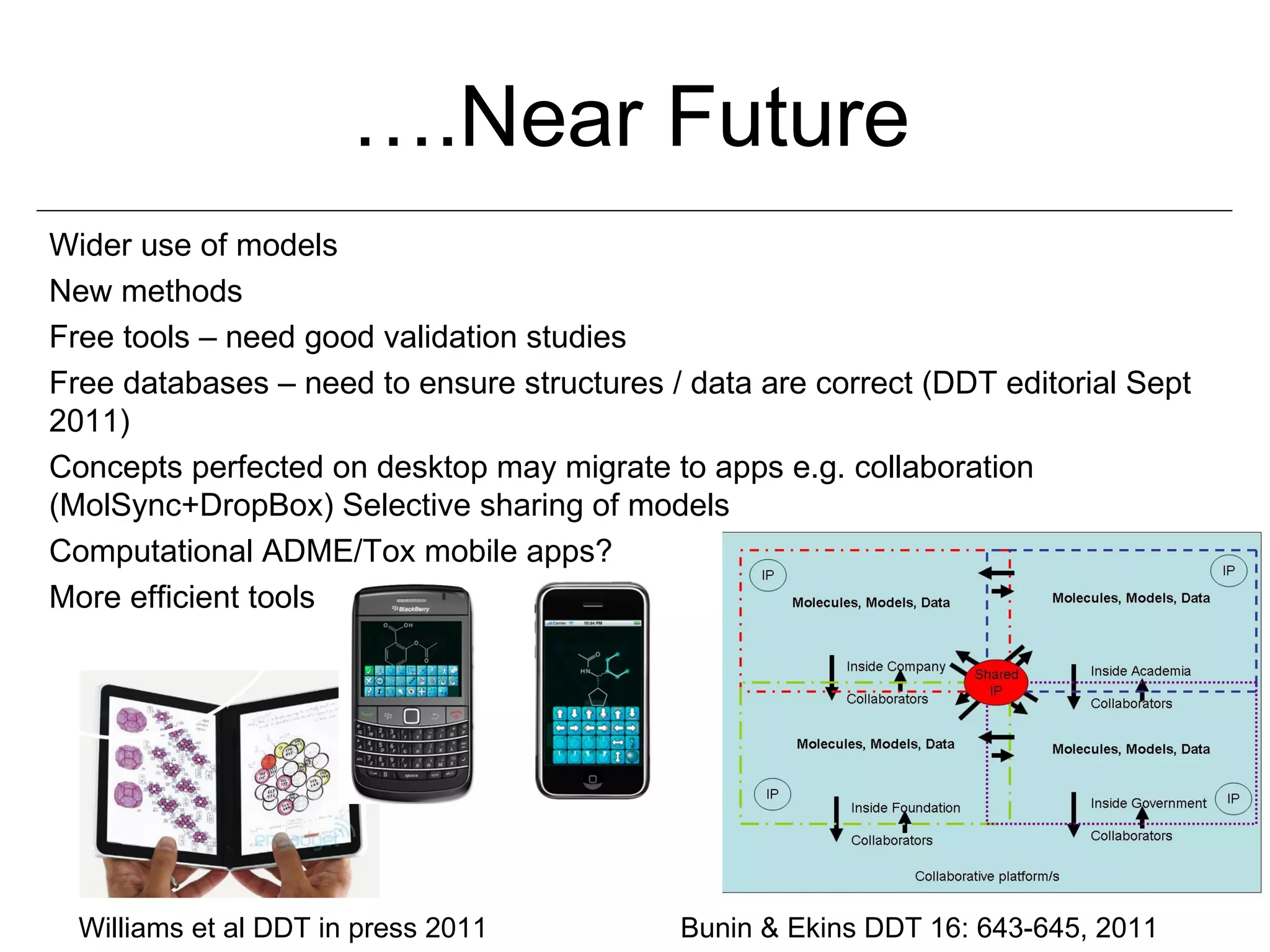


























Computational models are increasingly being used to predict human toxicities. Key enablers of these models include greater availability of data and open source tools. Models have been developed for physicochemical properties, various proteins like cytochromes and transporters, and complex properties like mutagenicity. Future areas of modeling include mitochondrial toxicity, more transporters, nuclear receptors, and green chemistry applications. Wider use of validated open source models and databases is expected, along with mobile applications and more efficient collaboration tools.

















































































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)





