Downloaded 81 times



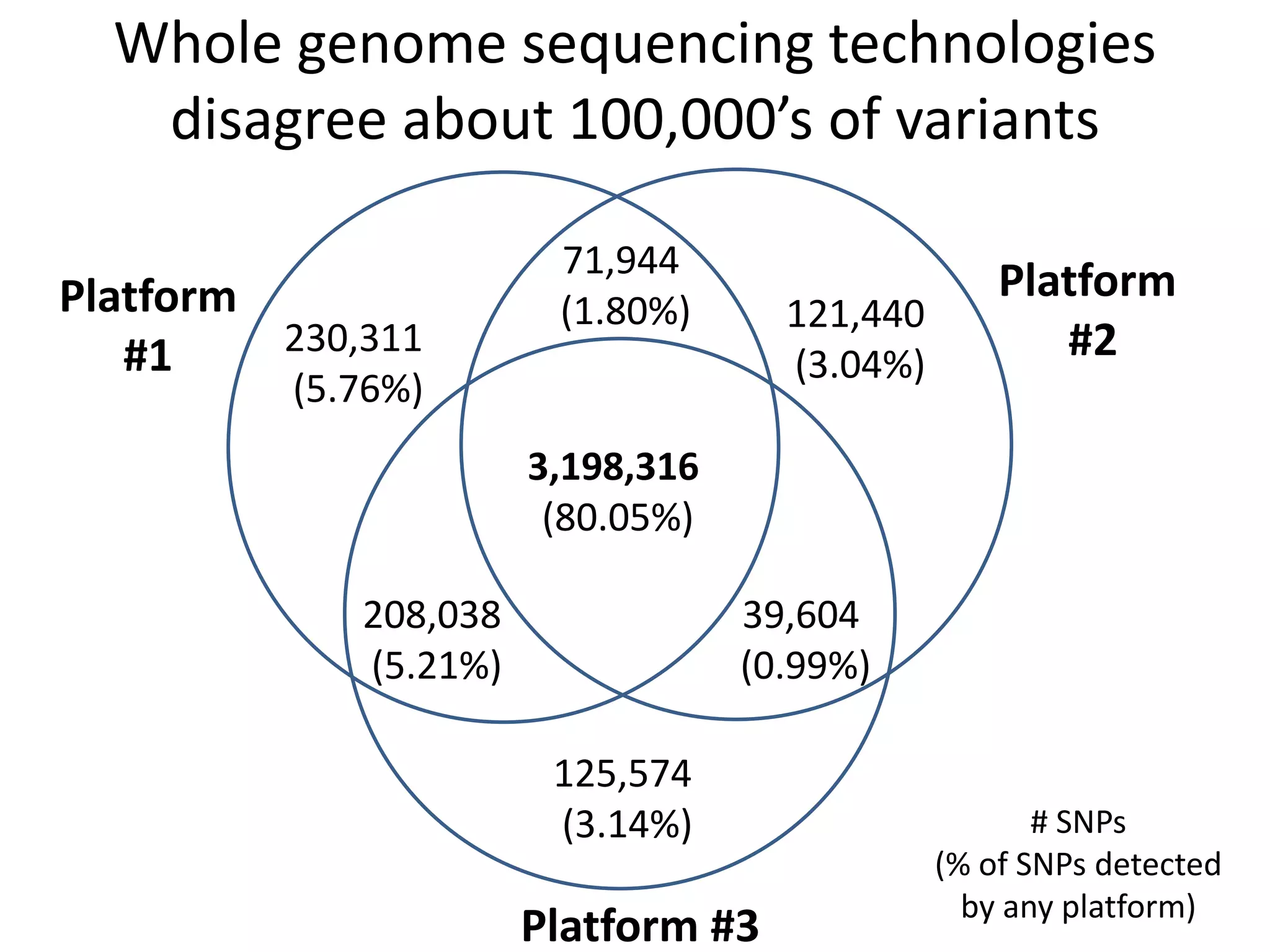

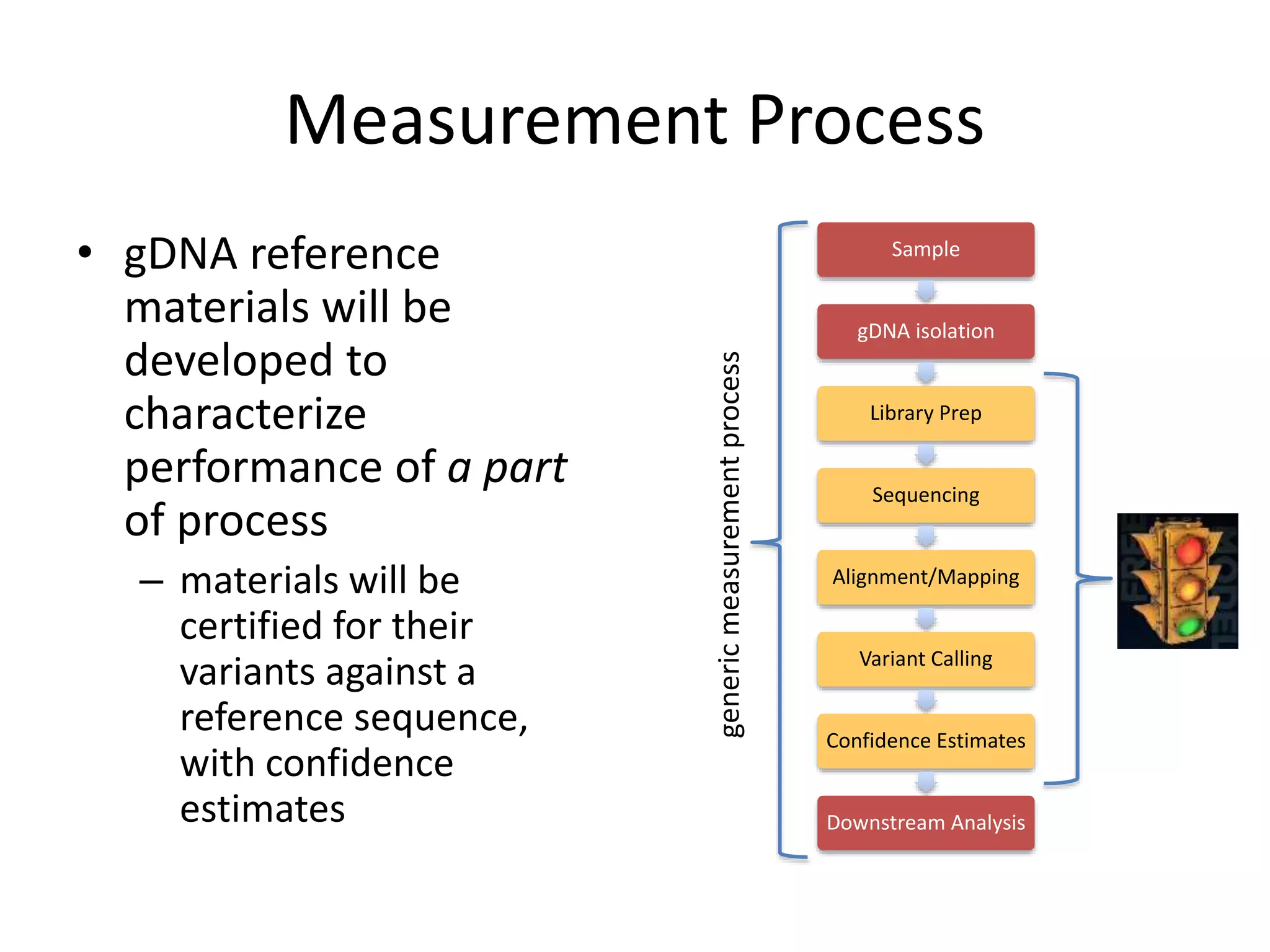




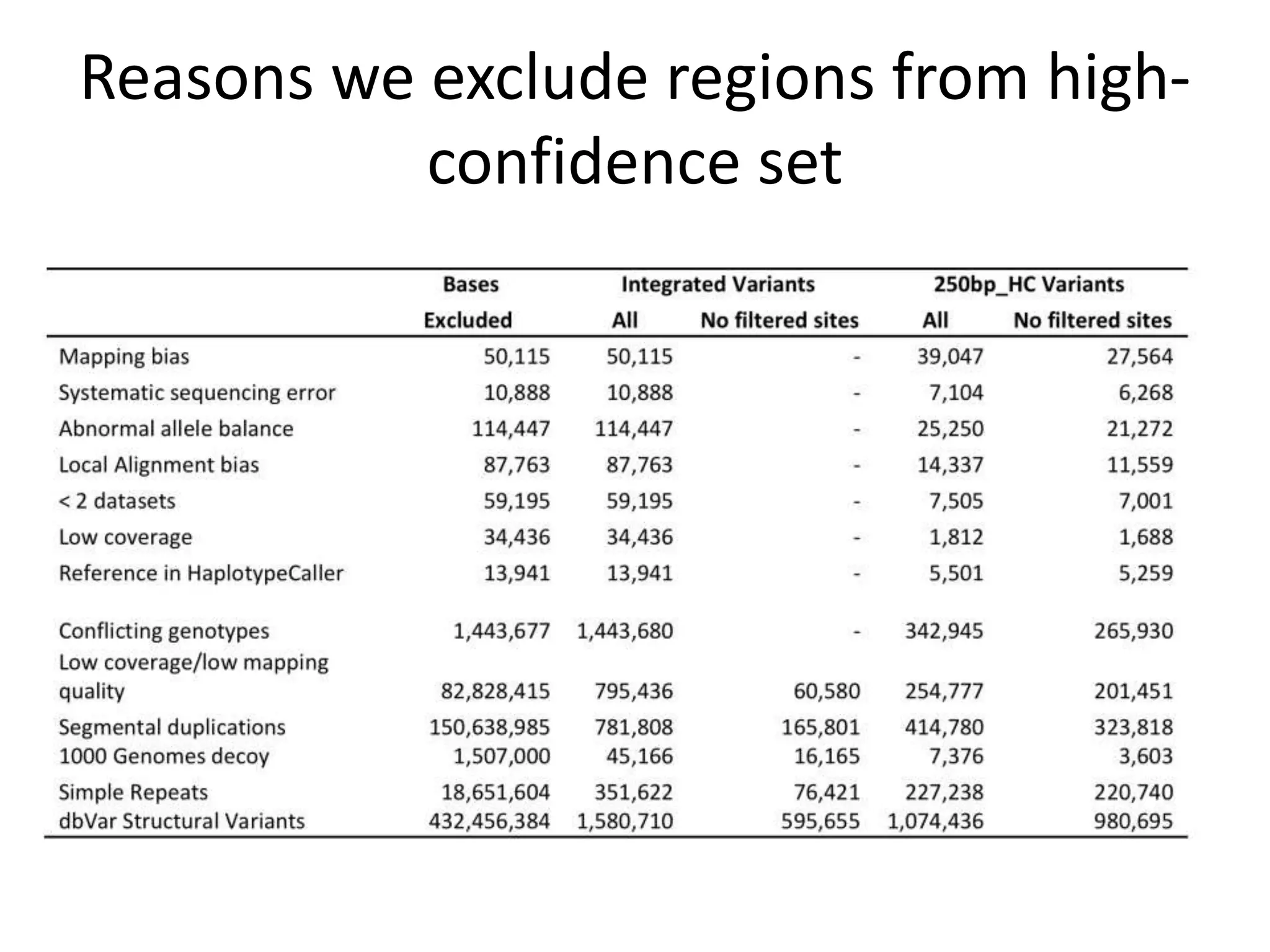



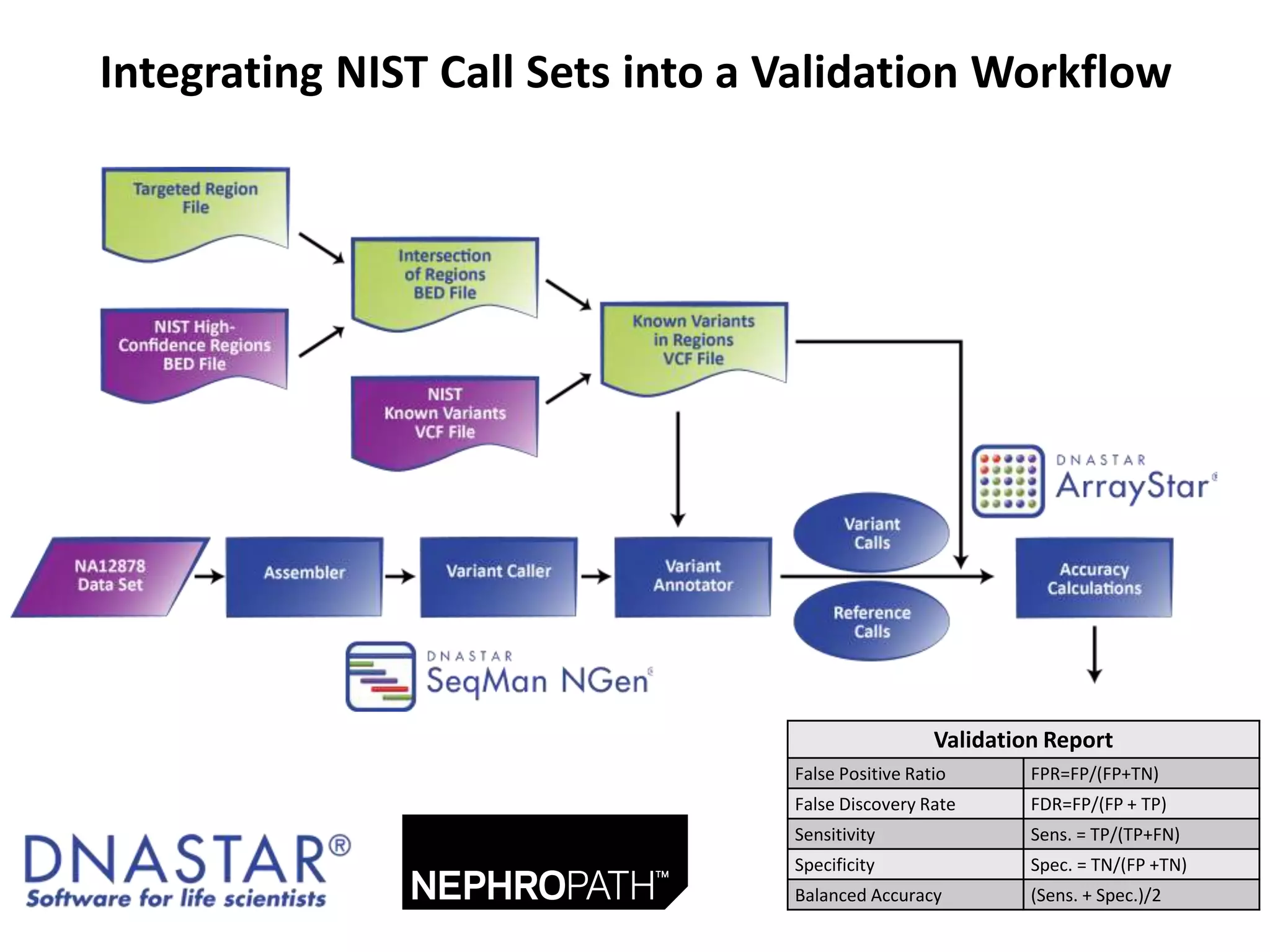
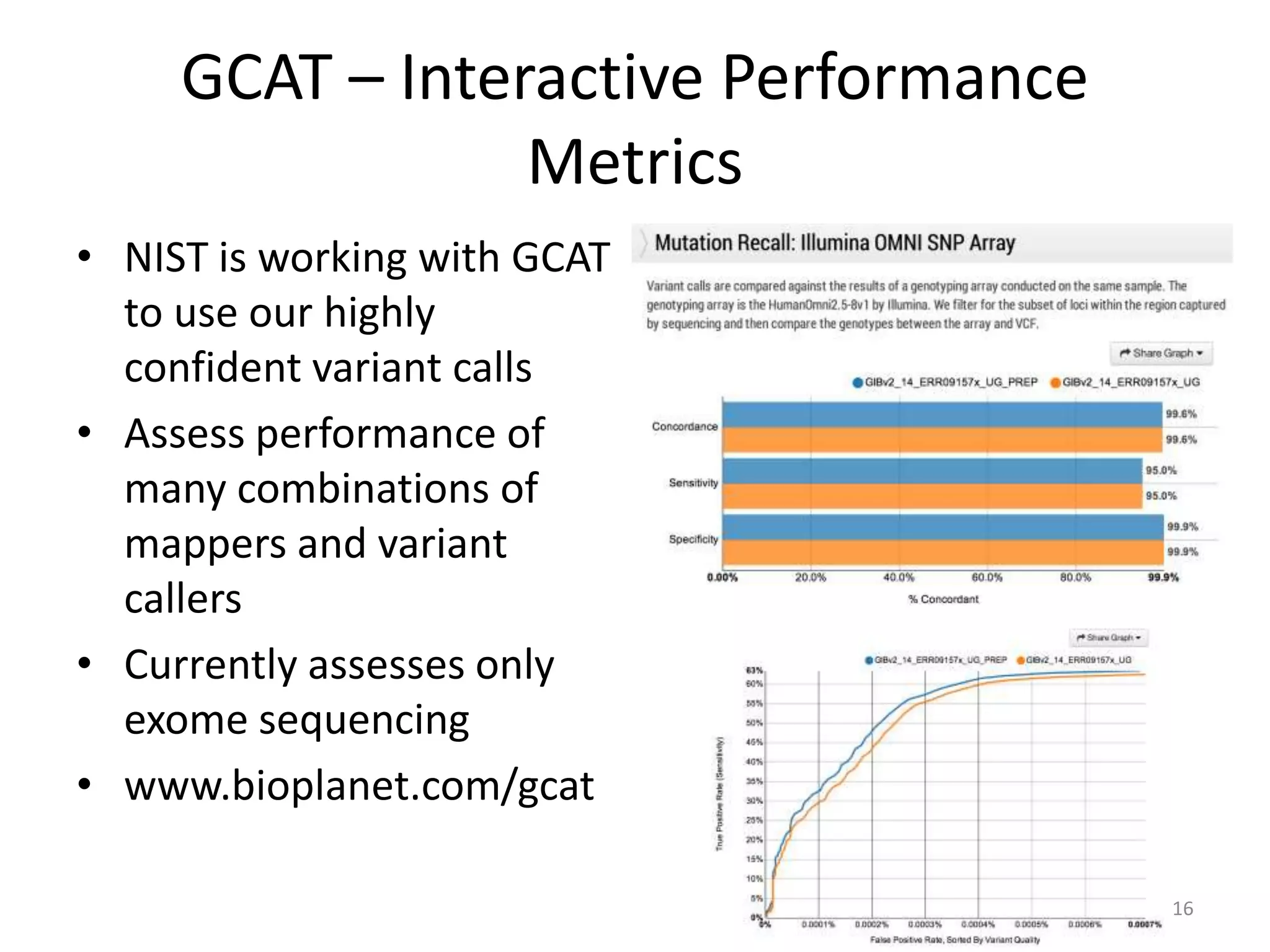

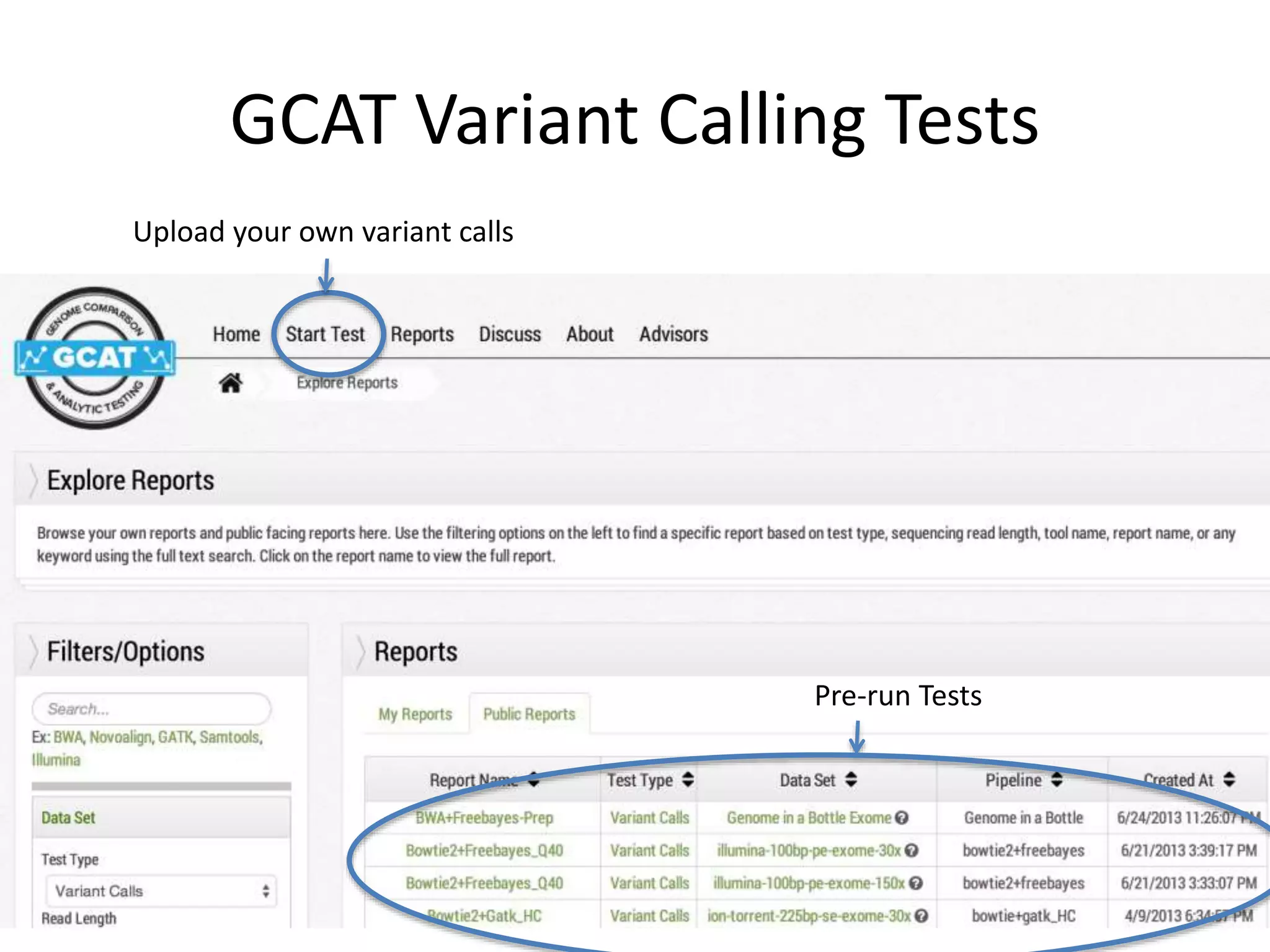
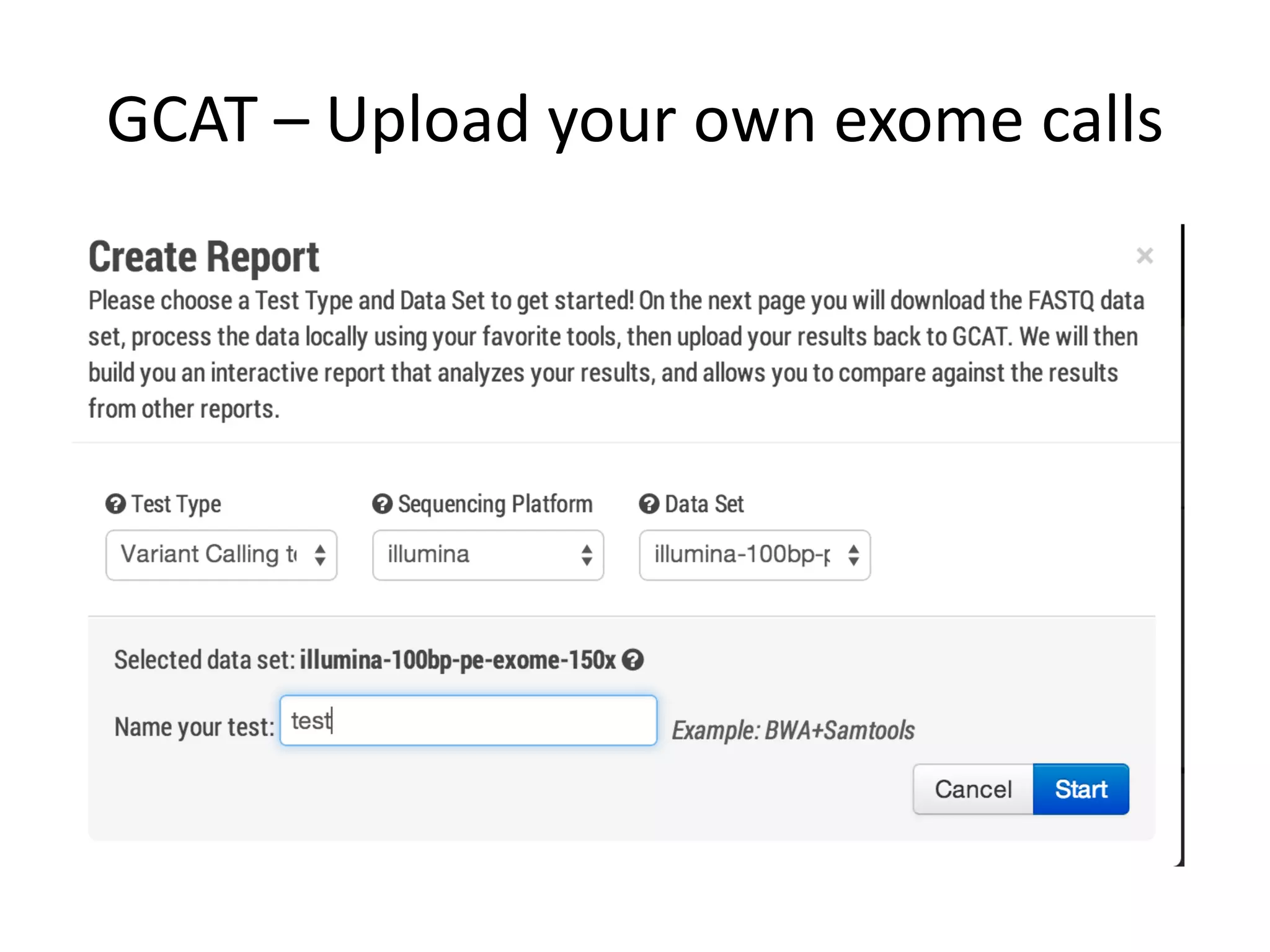
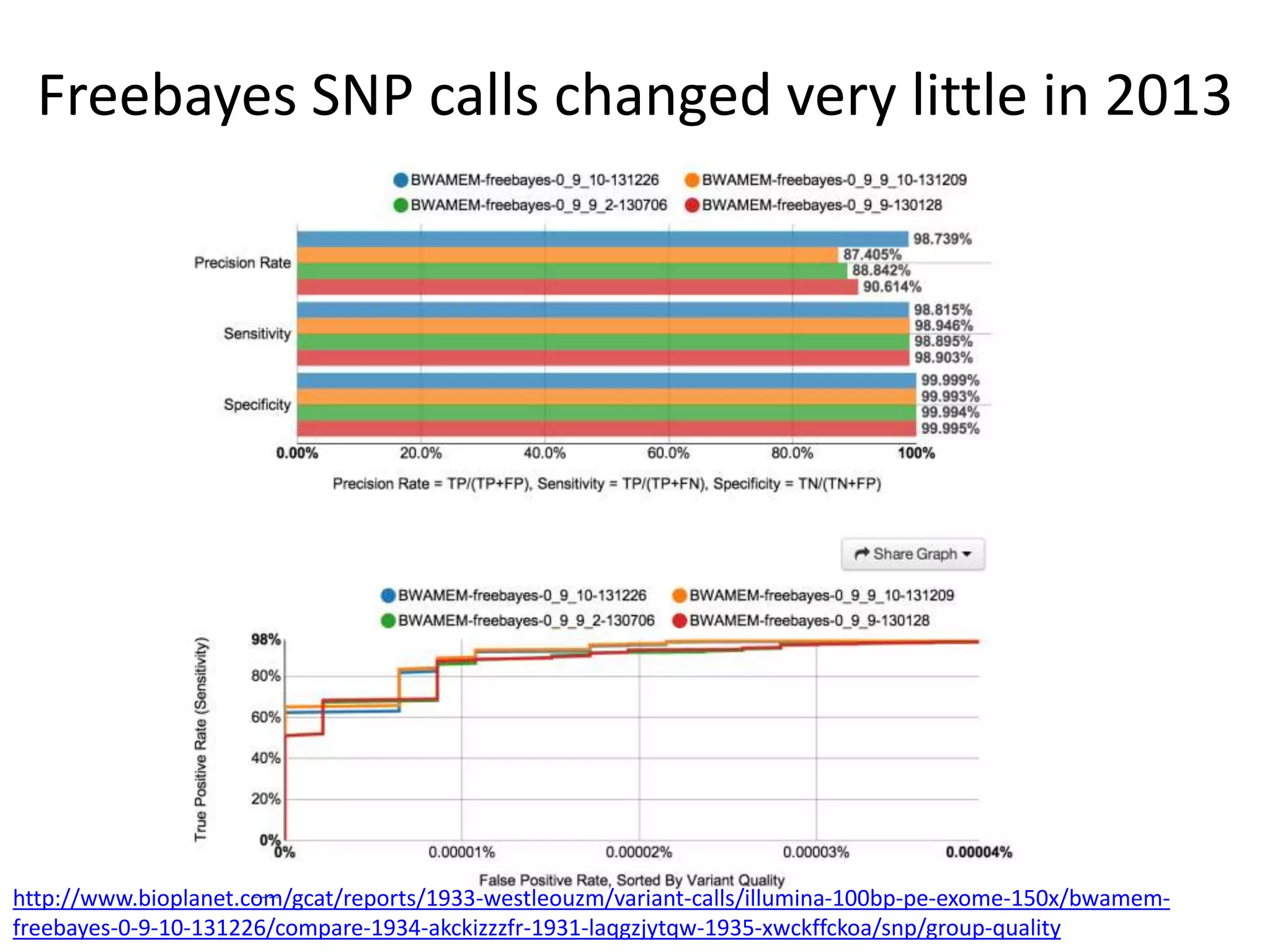
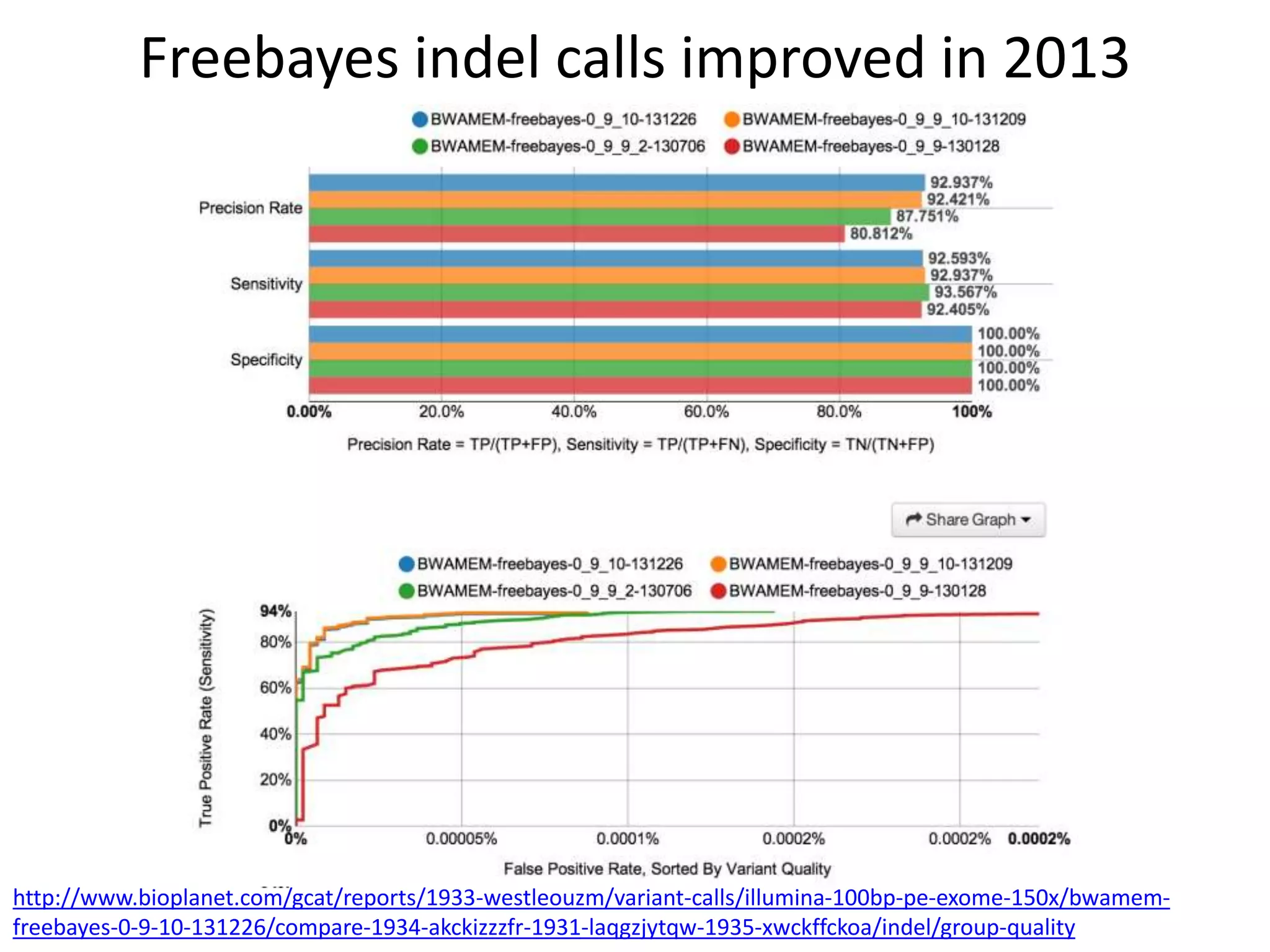


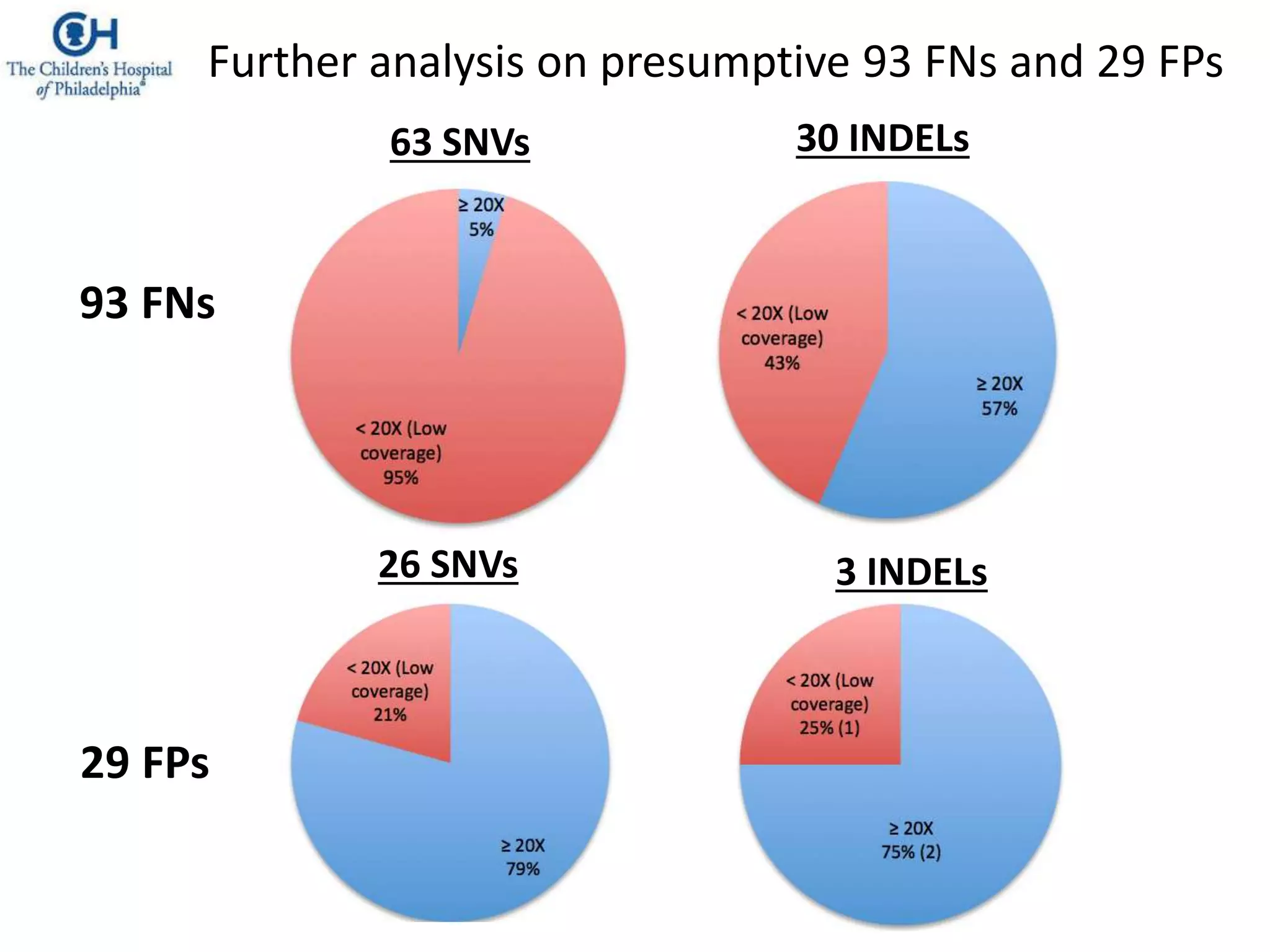
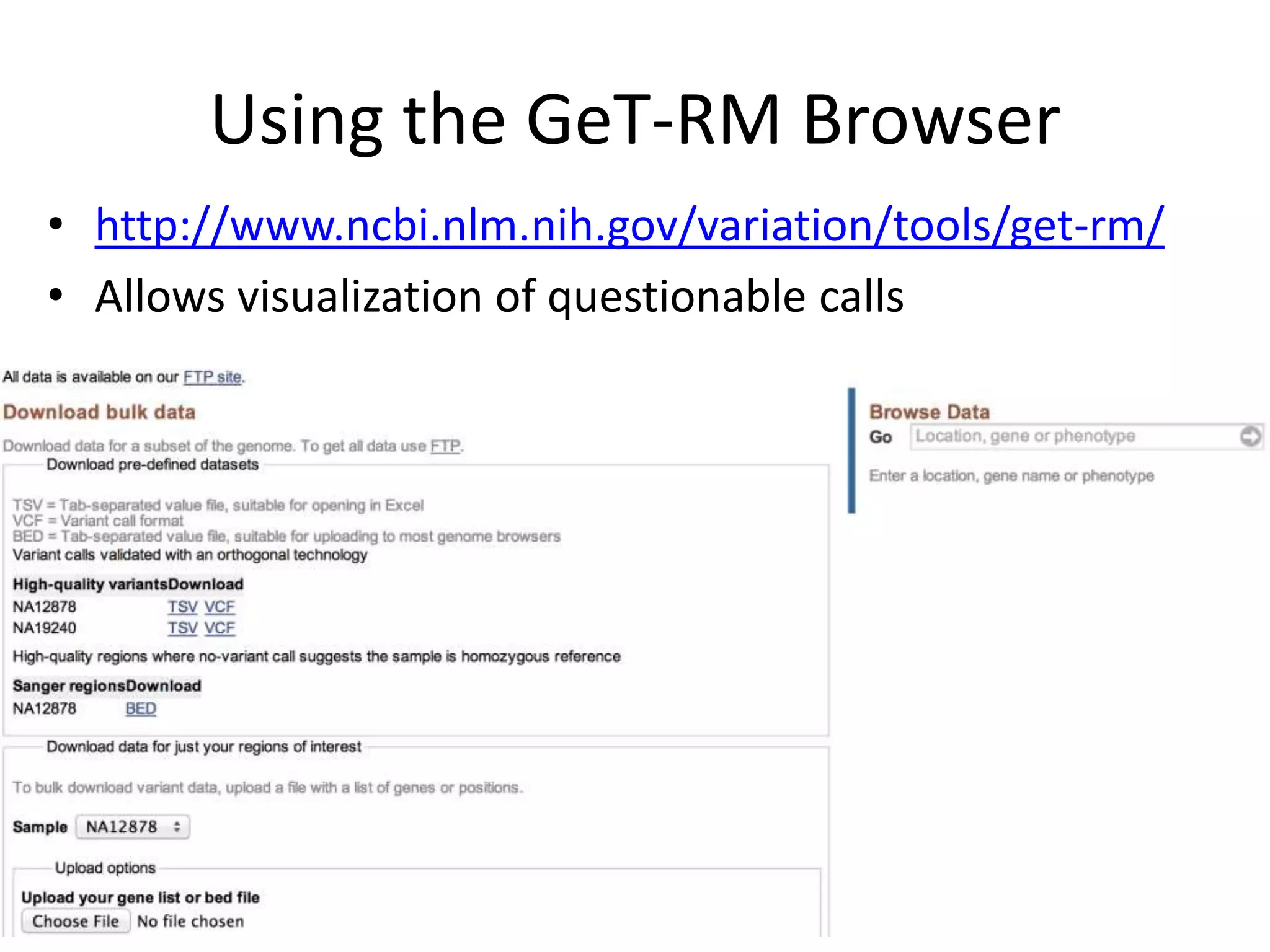
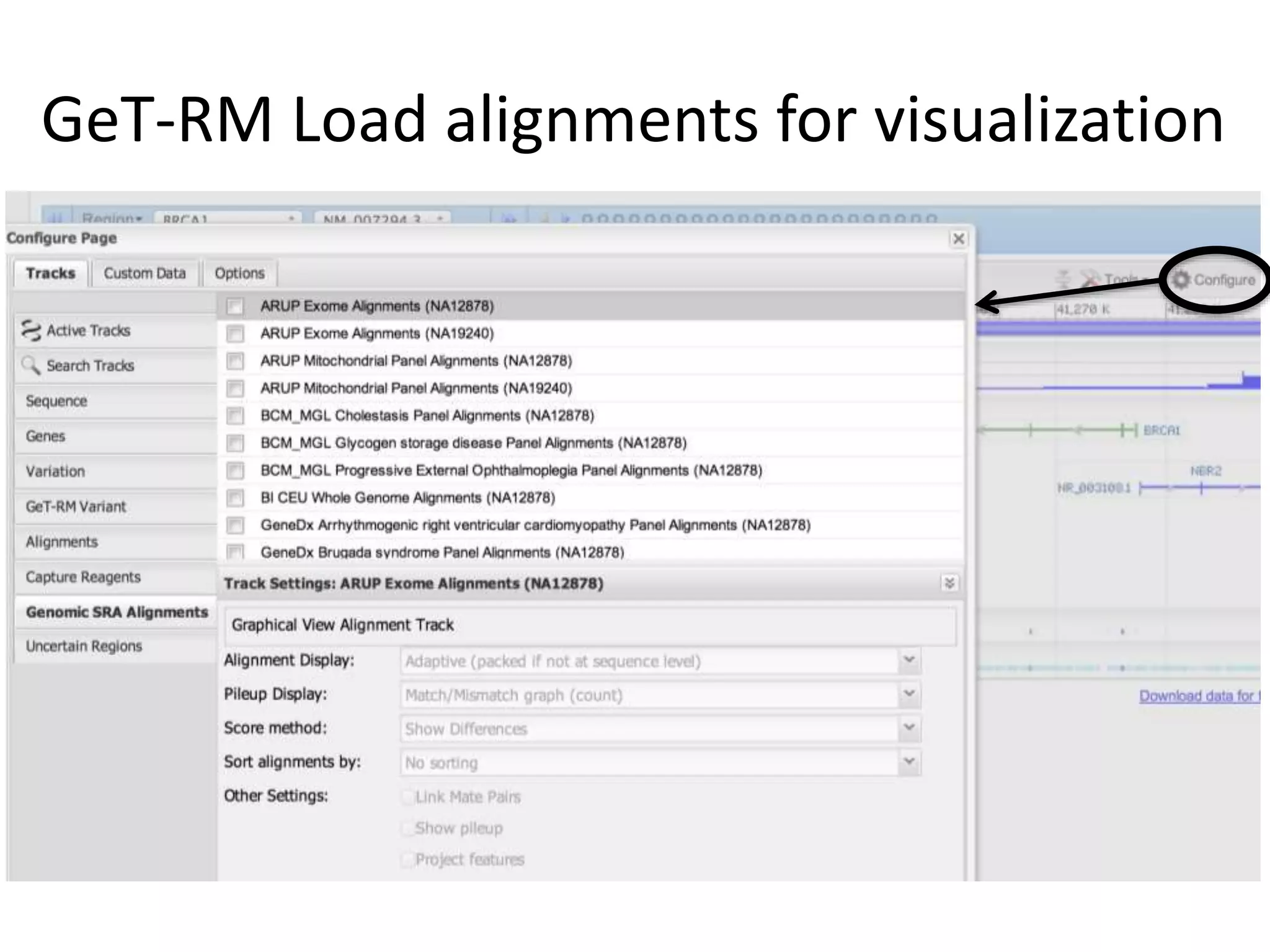
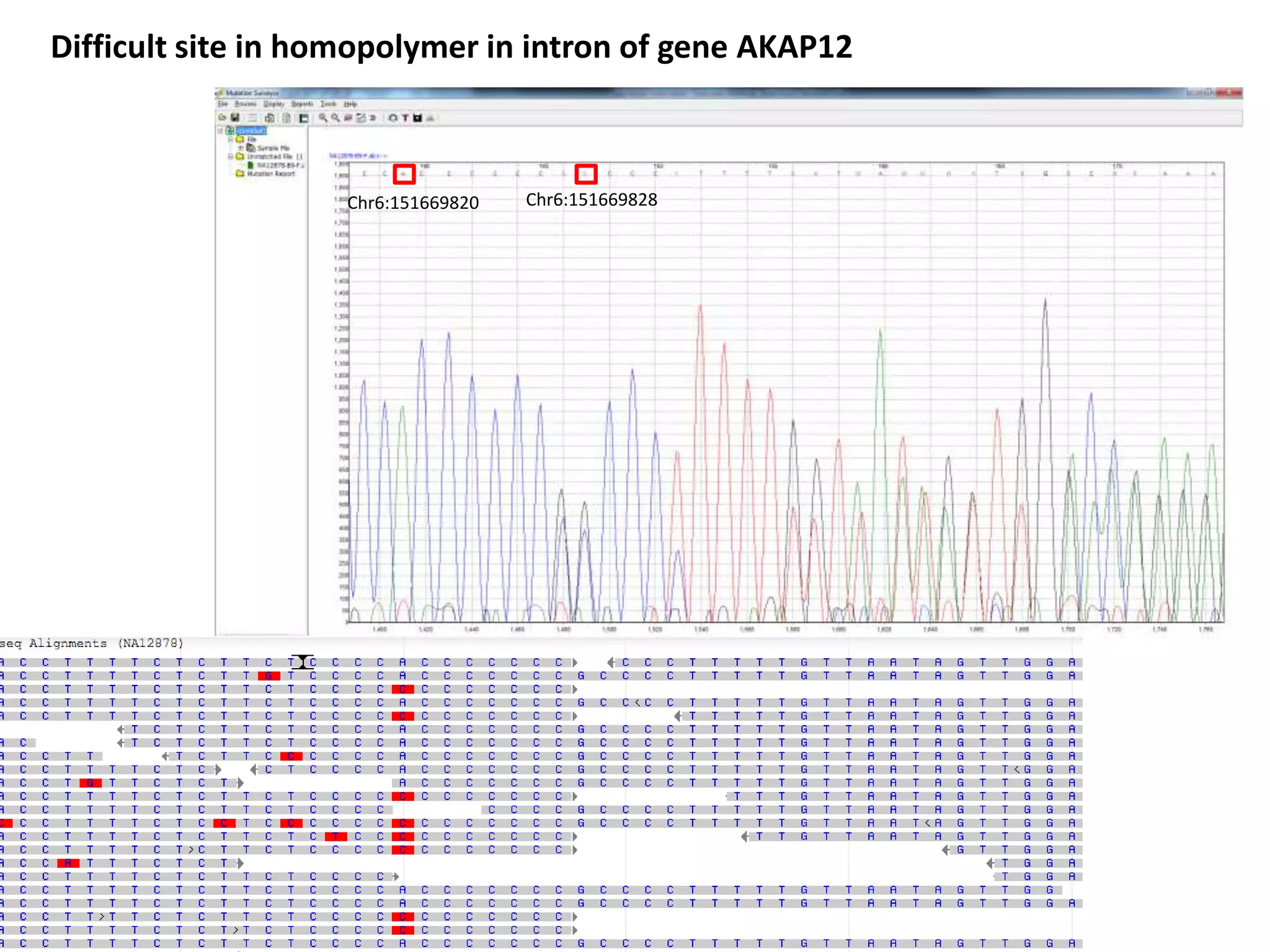
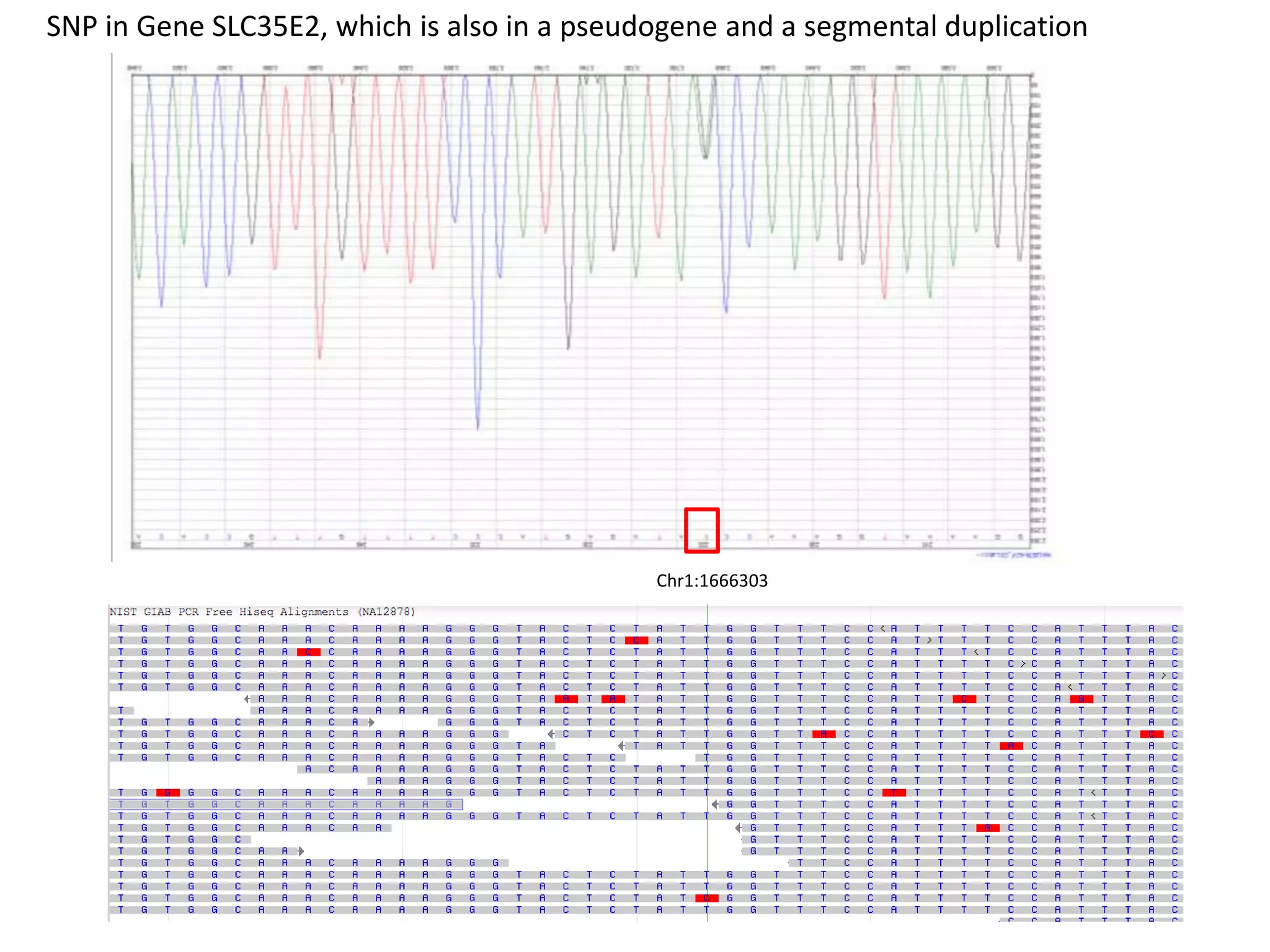
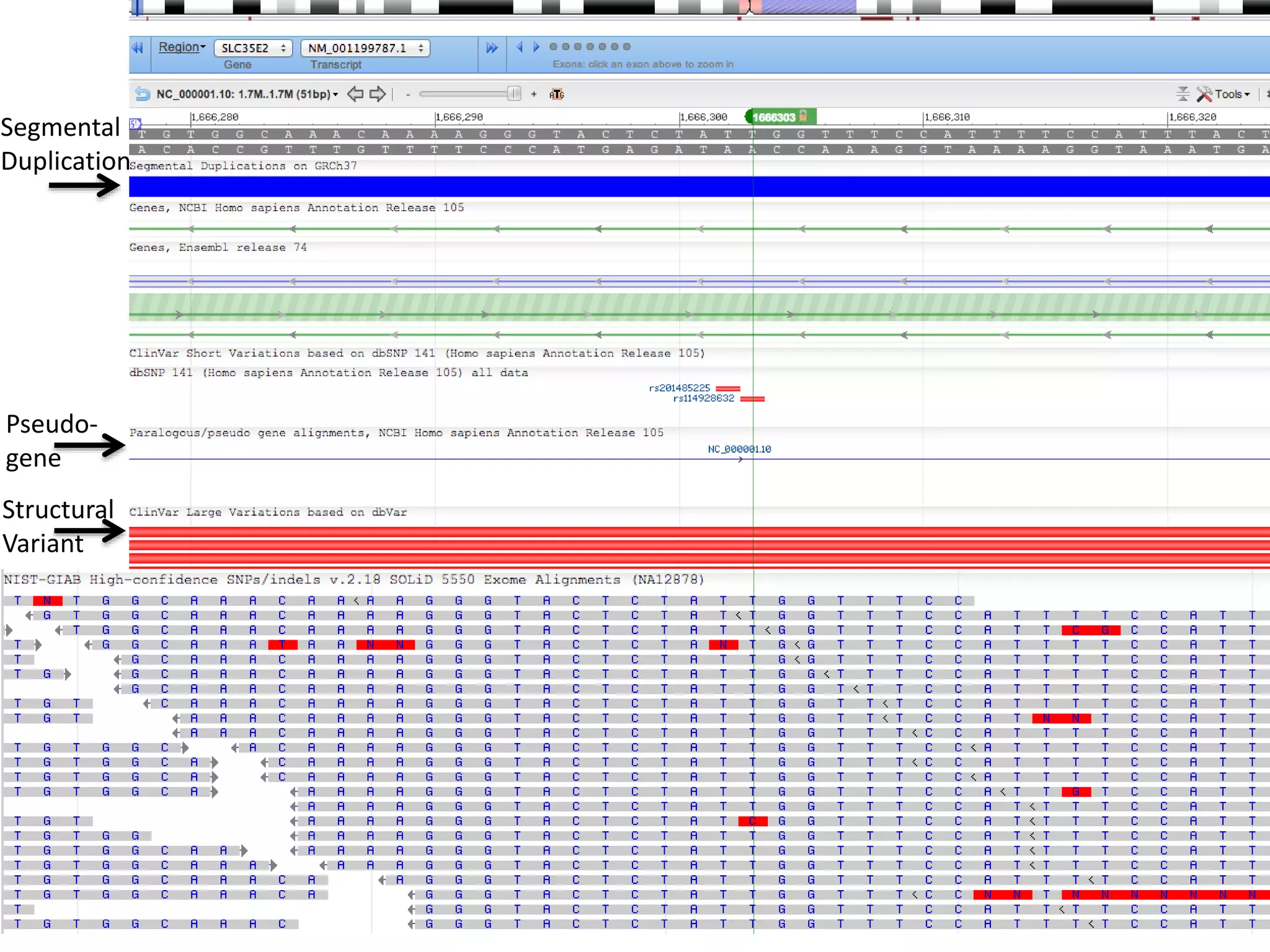

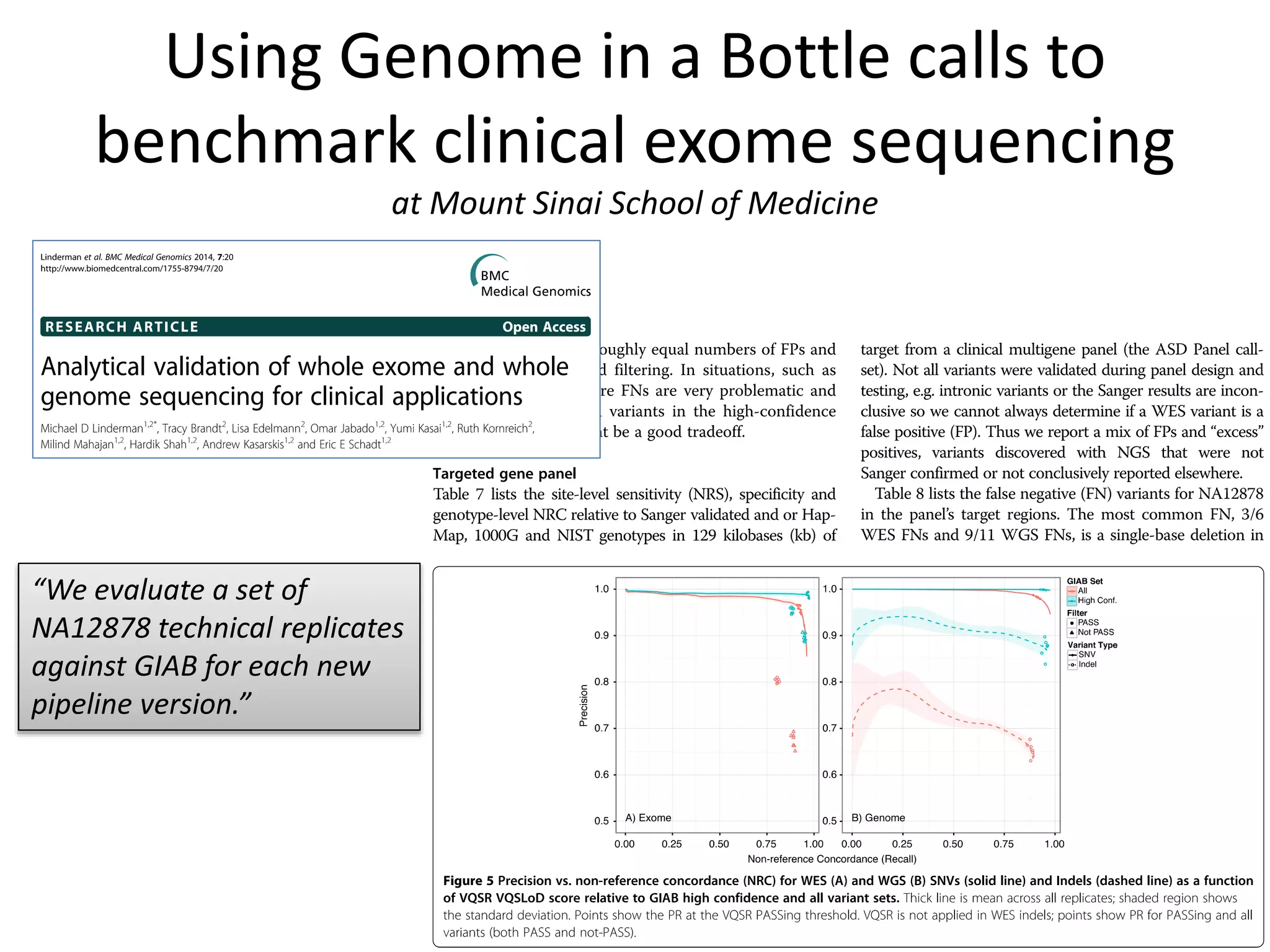

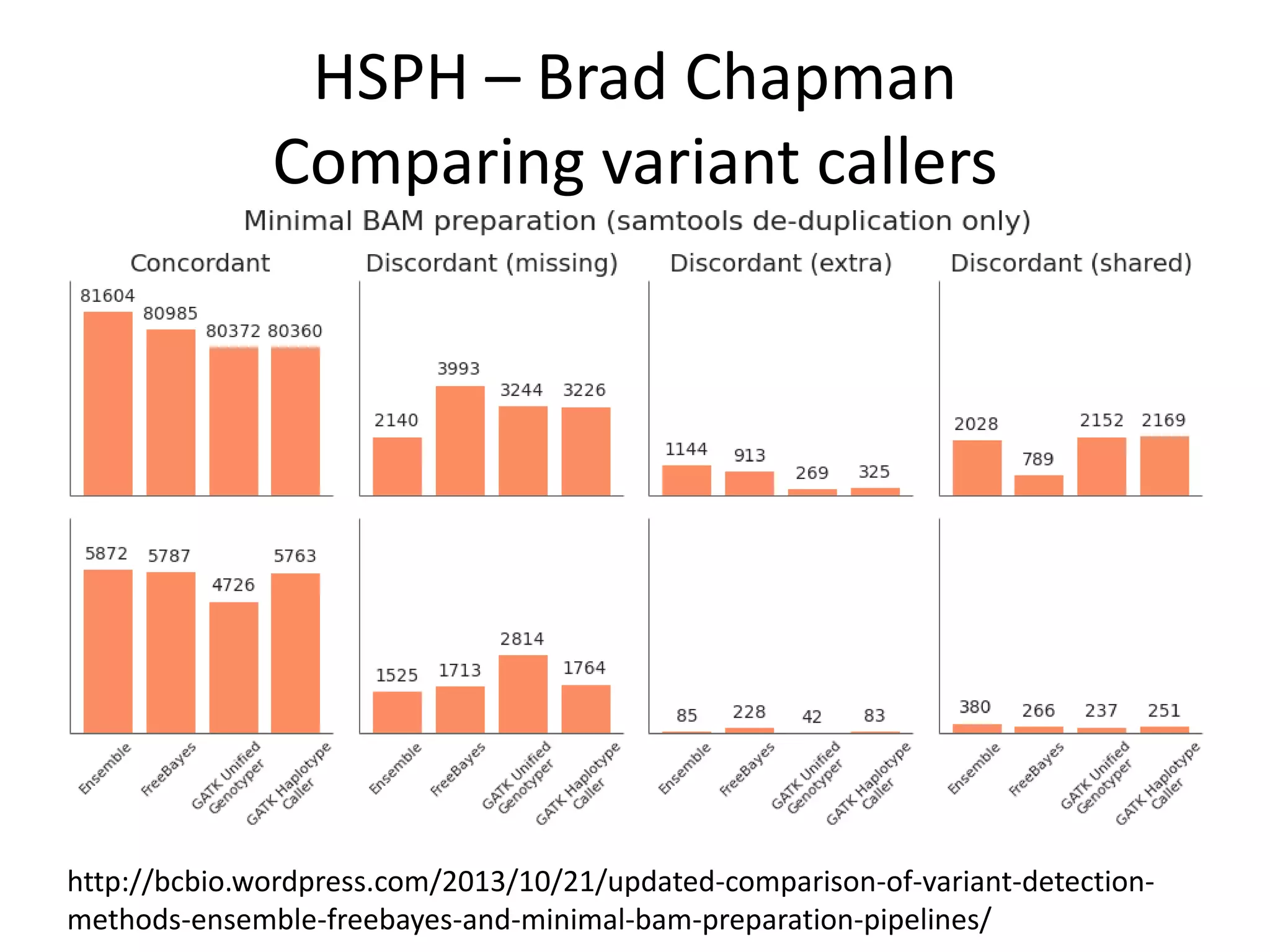
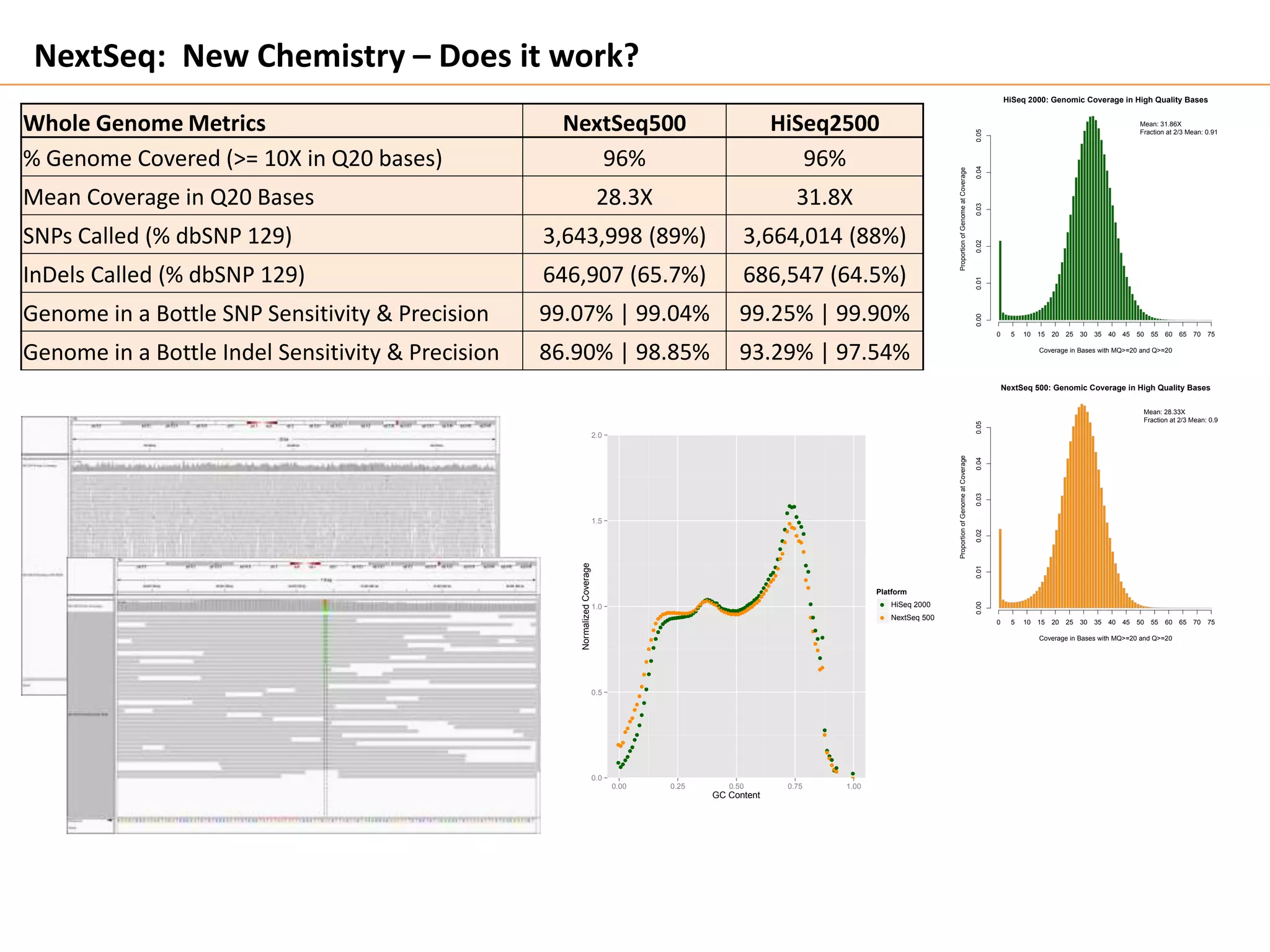

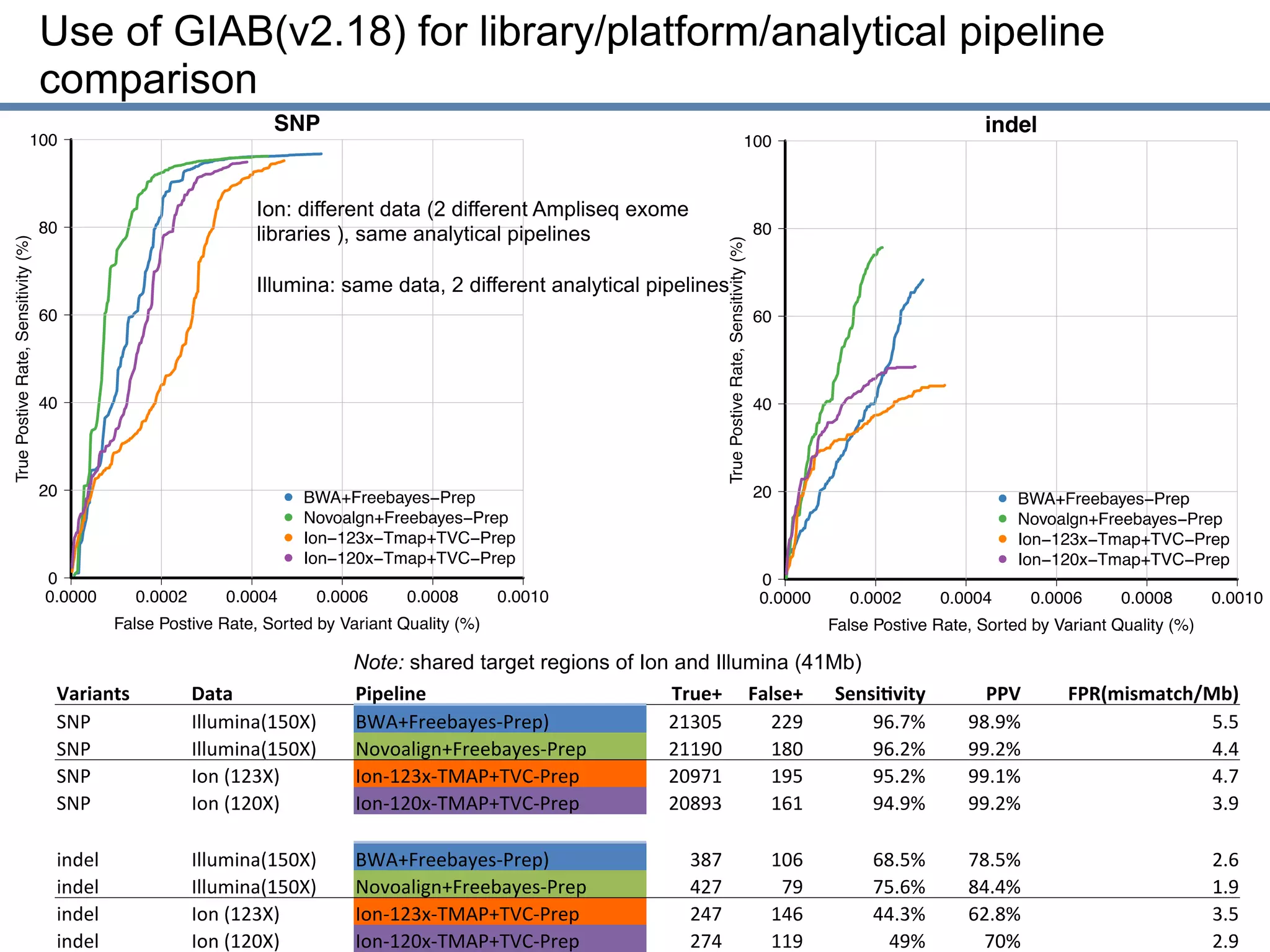









































This document discusses the Genome in a Bottle Consortium's efforts to develop reference materials and standards to validate next generation sequencing assays. It provides an overview of the consortium's goals to generate reference genomes with highly confident variant calls and accompanying data to allow labs to compare results and assess false positives and false negatives. The document describes some examples of how labs are using the consortium's data on the NA12878 genome to benchmark sequencing platforms and bioinformatics workflows.










































































![Rheumatic Fever CASE PRESENTATION [Autosaved].pptx](https://cdn.slidesharecdn.com/ss_thumbnails/casepresentationautosaved-251123182512-9d9b0da4-thumbnail.jpg?width=640&height=640&fit=bounds)











