Downloaded 62 times

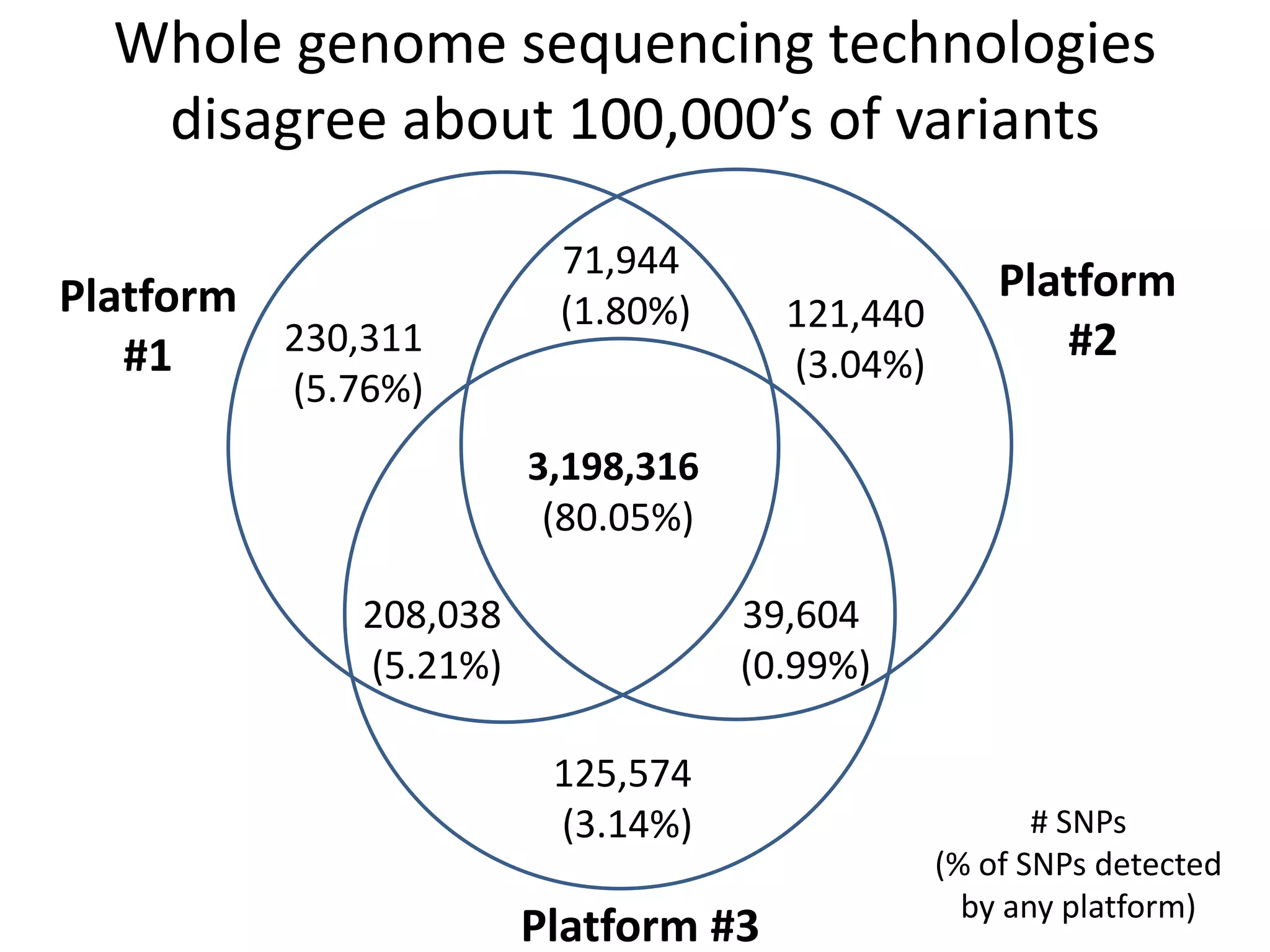





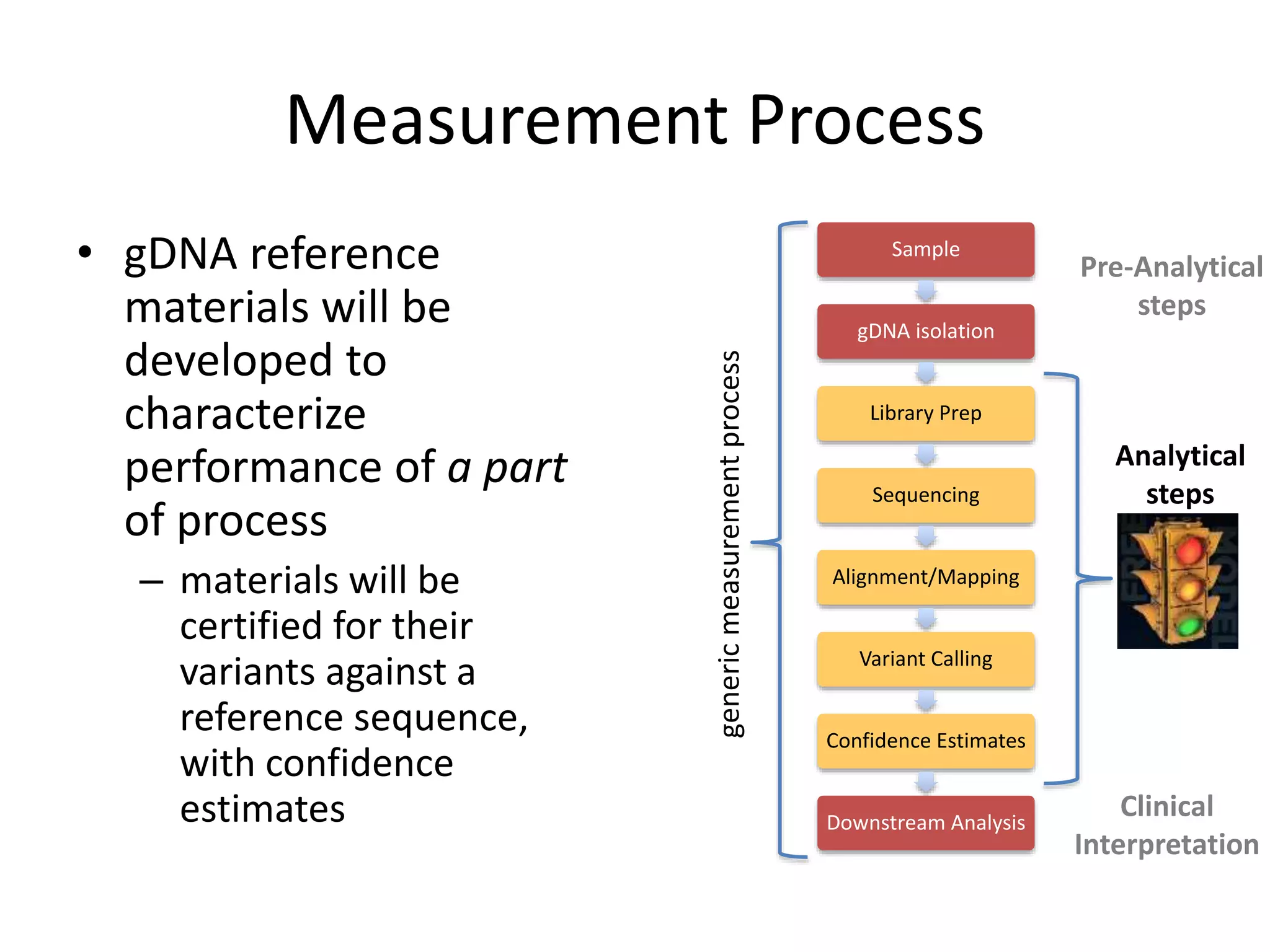
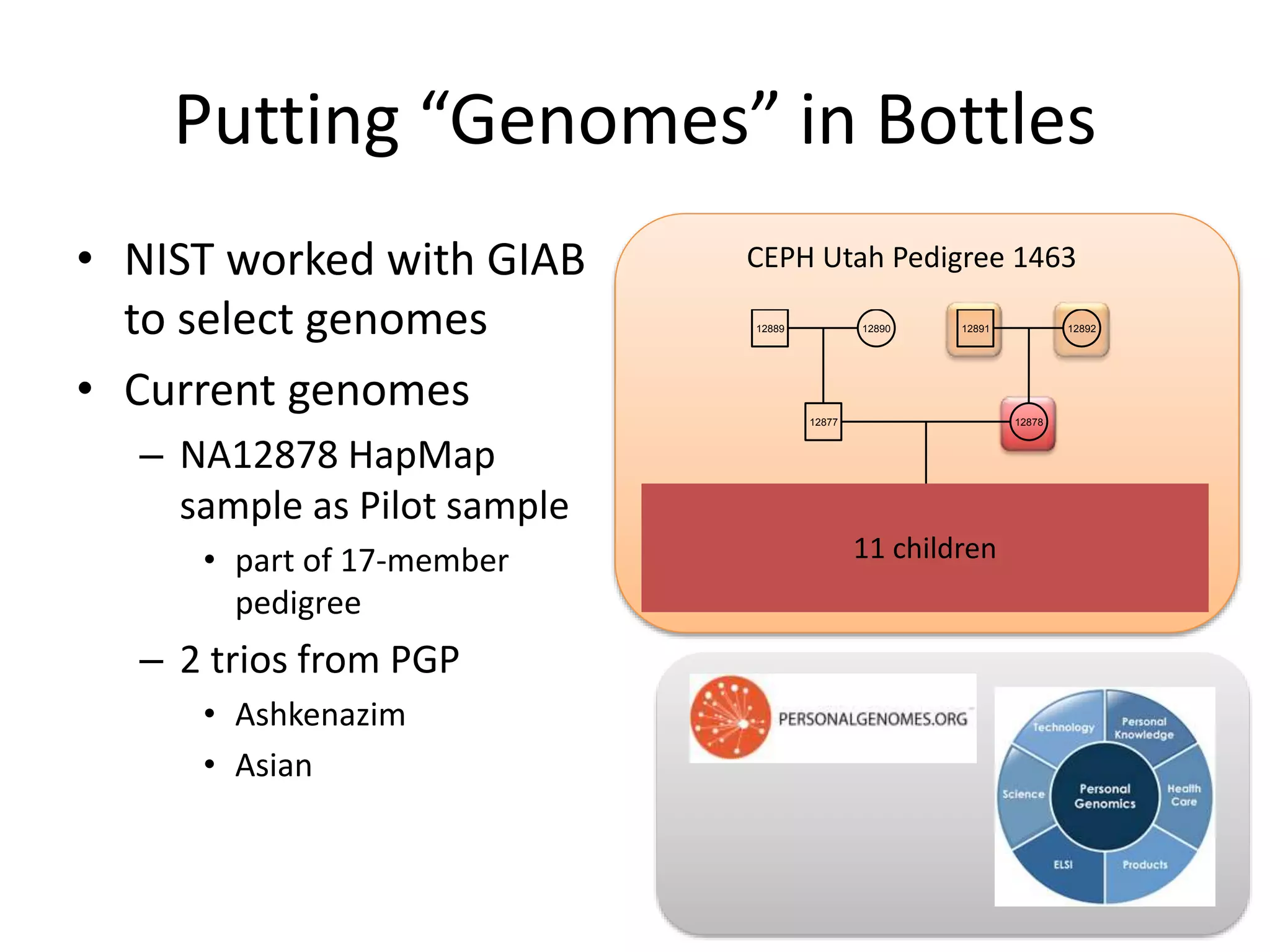


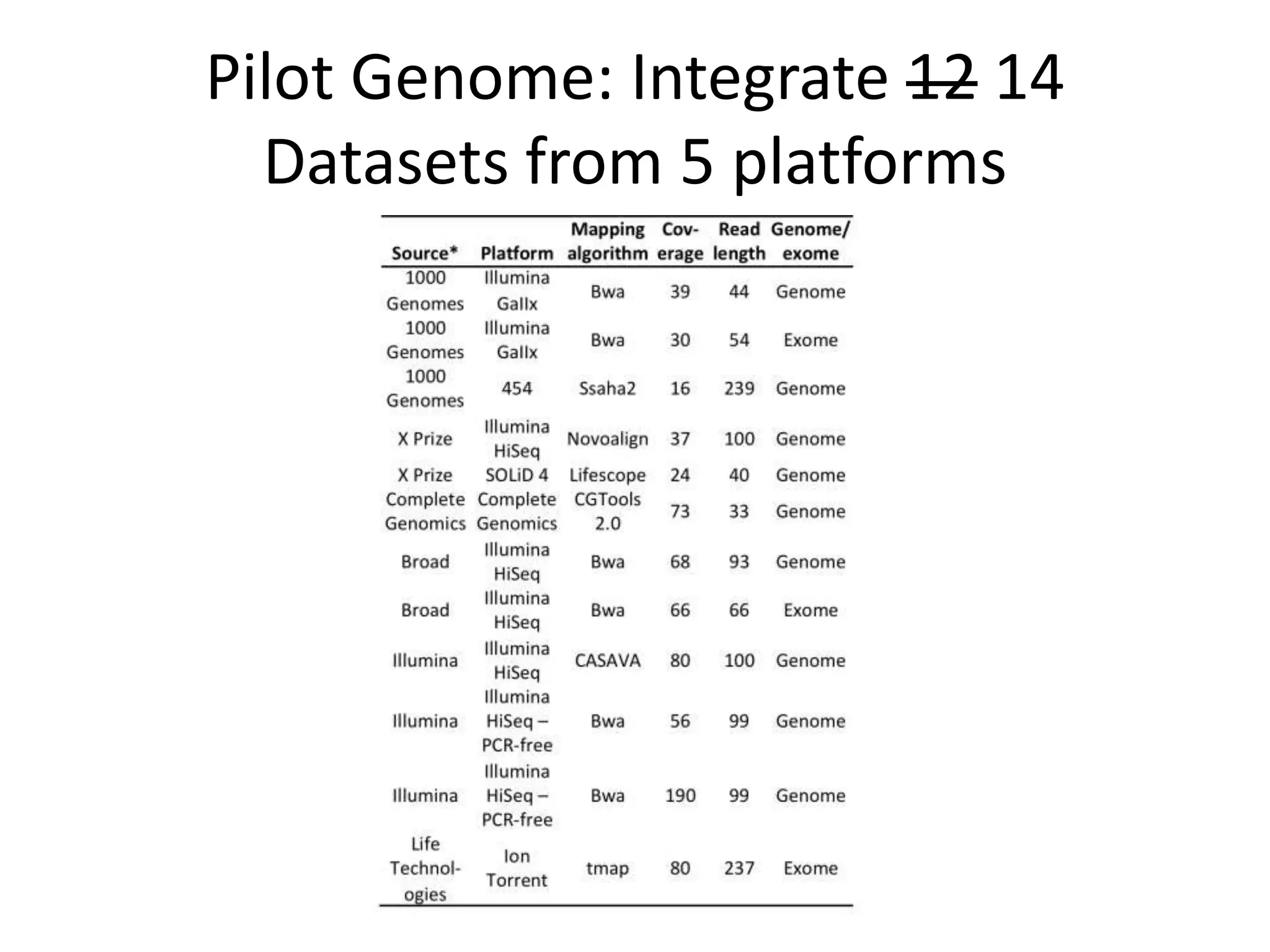
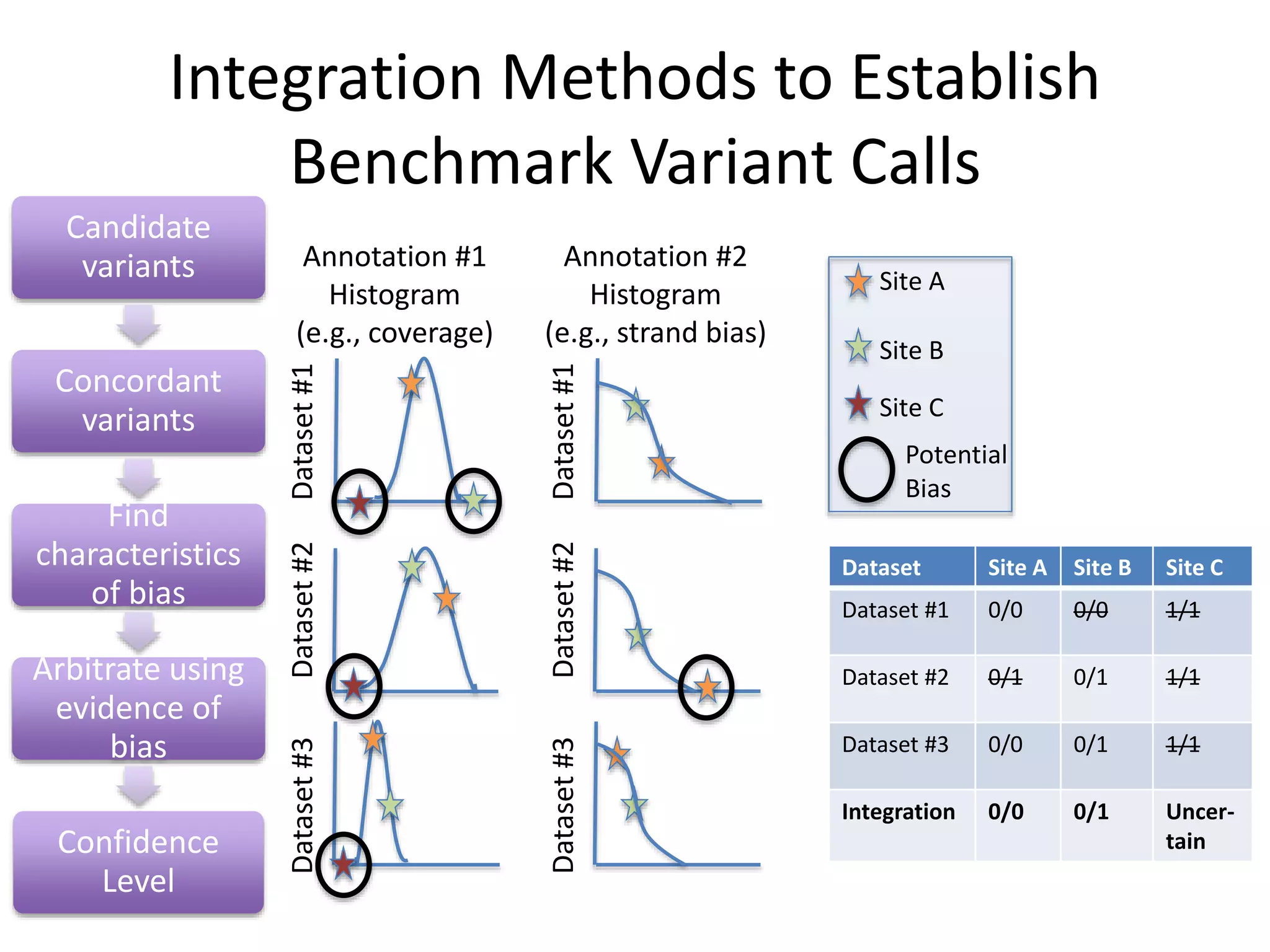



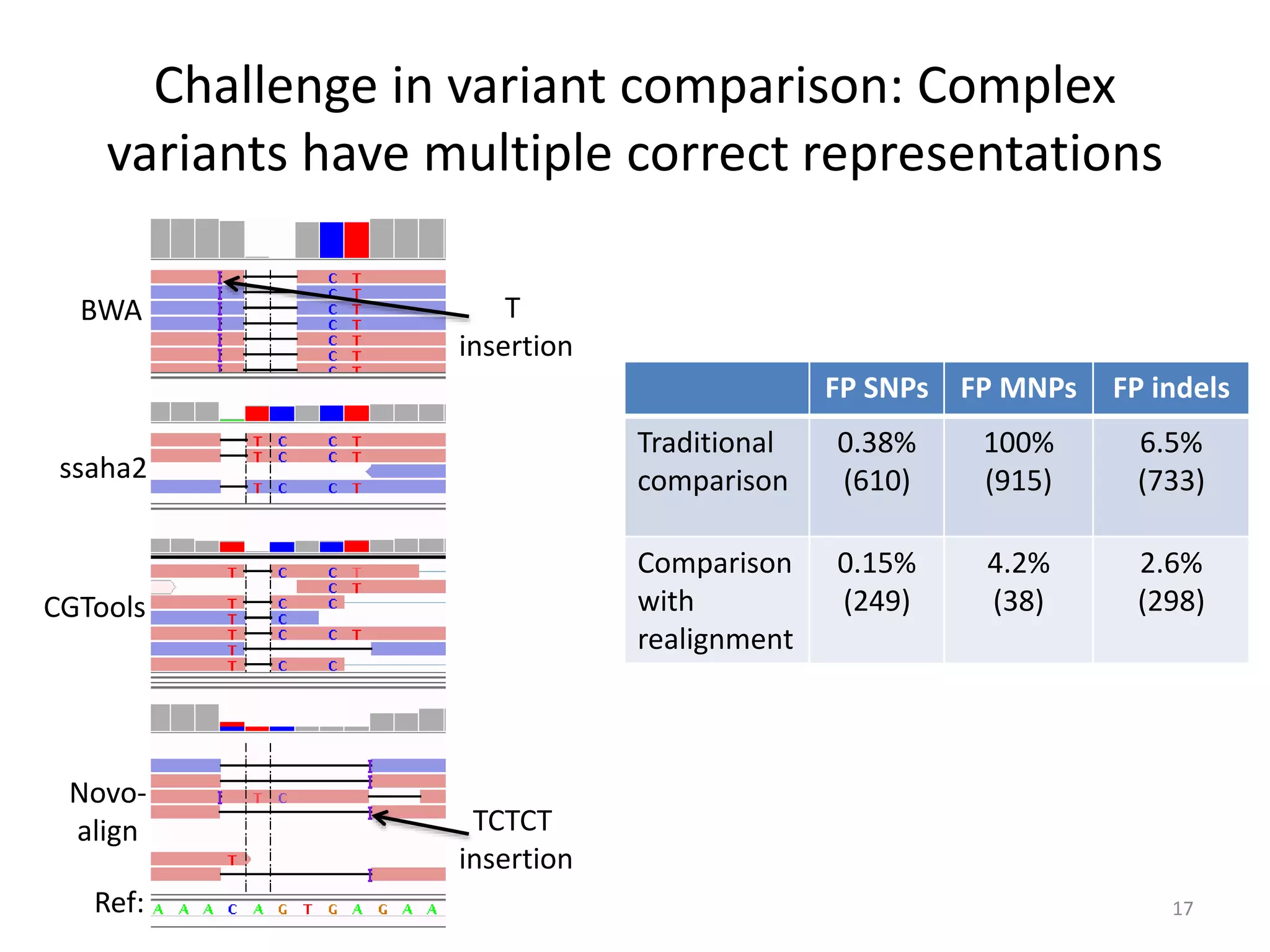
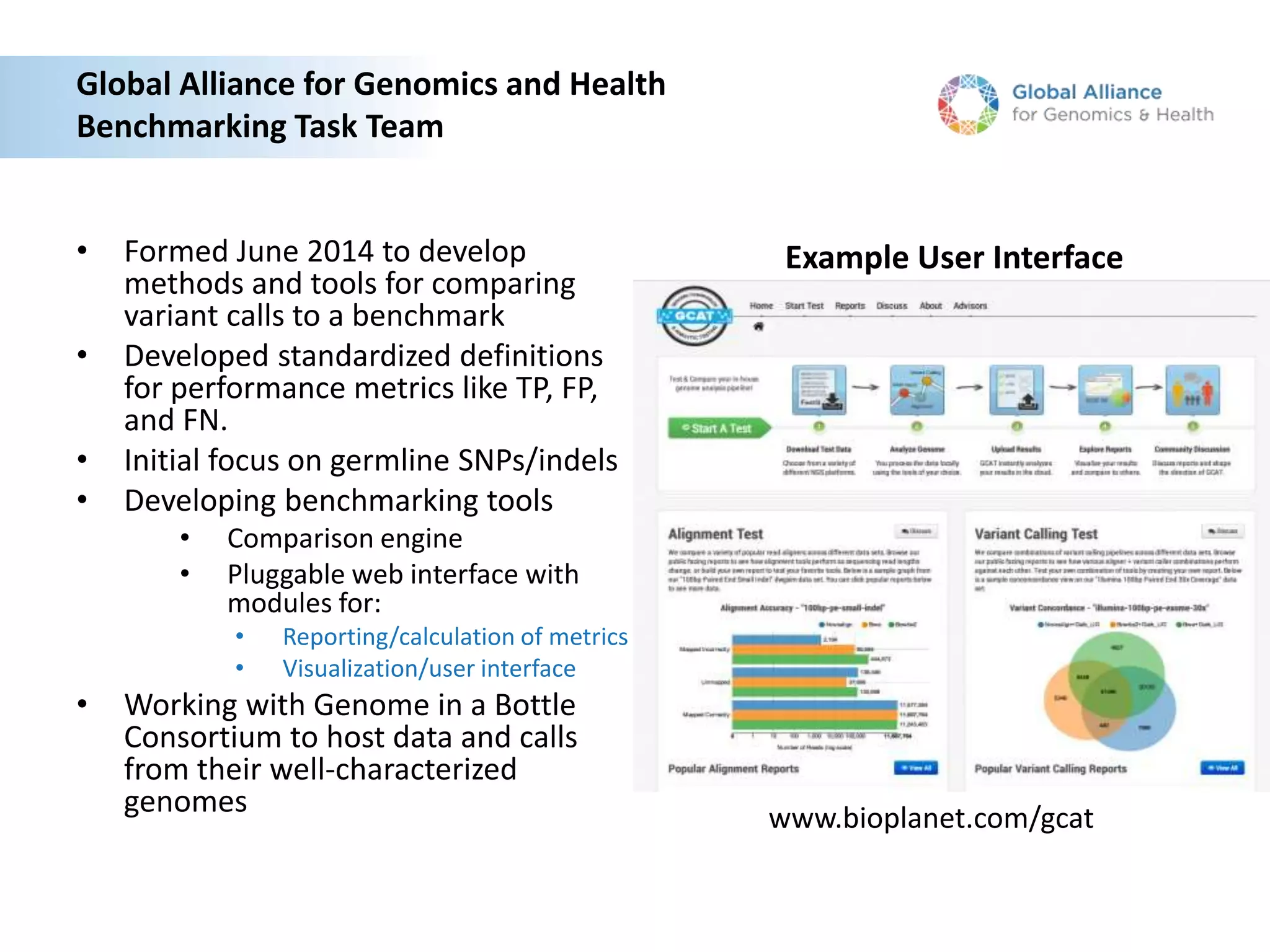

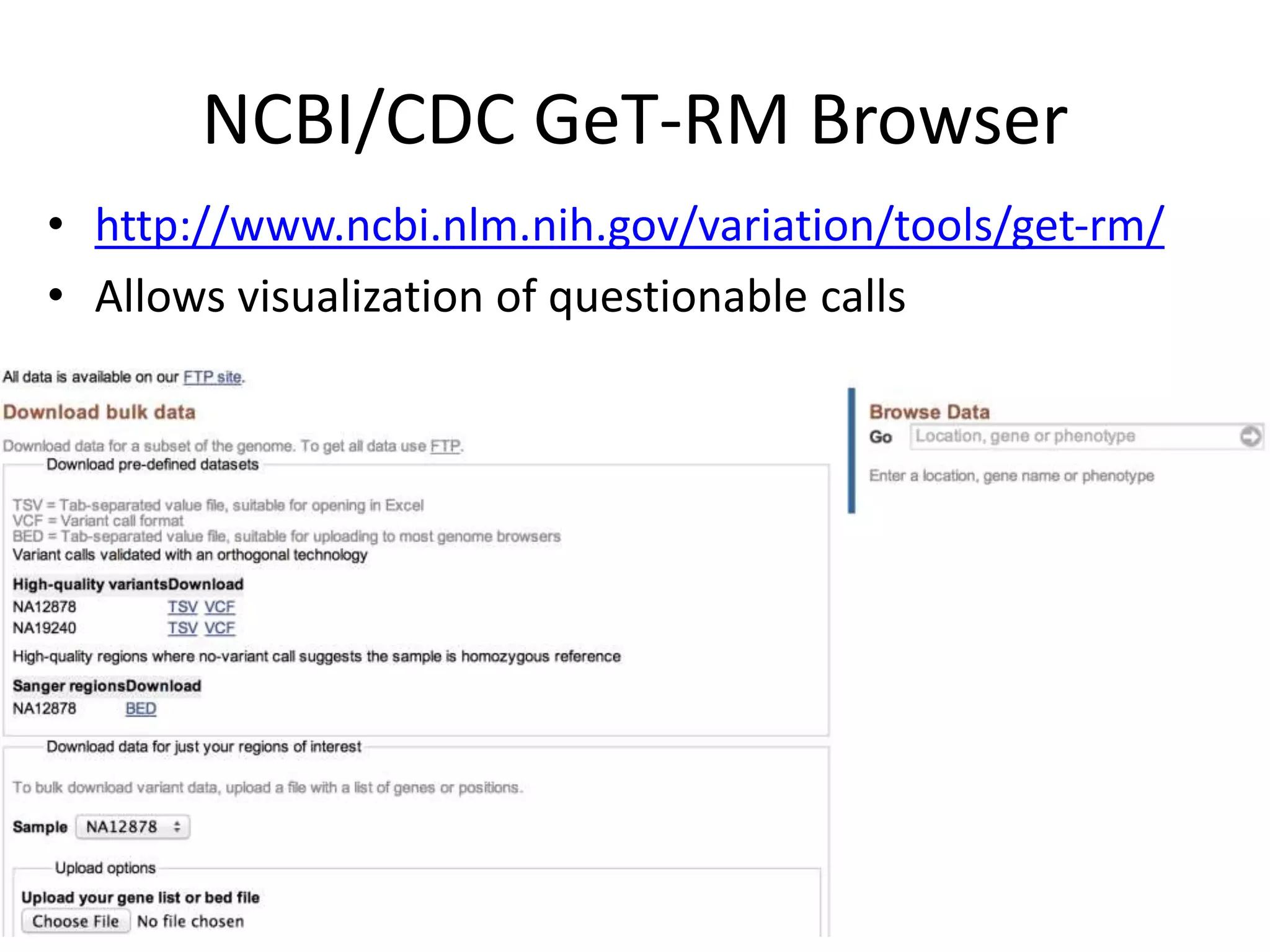





































This document summarizes the work of the Genome in a Bottle Consortium to develop reference materials for evaluating the accuracy of whole genome sequencing. It finds that different sequencing platforms and bioinformatics programs often disagree on called variants. The Consortium is developing well-characterized human genomes that will serve as standards, including the genome of individual NA12878. The goal is to assign confidence levels to called variants and provide integrated variant calls across multiple datasets to help benchmark sequencing accuracy and evaluate sequencing performance in different genomic and clinical contexts.























































































