


















































































































































































































































































































The document discusses risk assessment in the pharmaceutical industry, focusing on the relationship between material attributes, process parameters, and critical quality attributes (CQAs). It outlines methodologies for risk assessment, quality risk management processes, and the significance of critical process parameters in ensuring product quality. Additionally, it emphasizes the importance of systematic risk management throughout the product lifecycle, from development to post-approval changes.
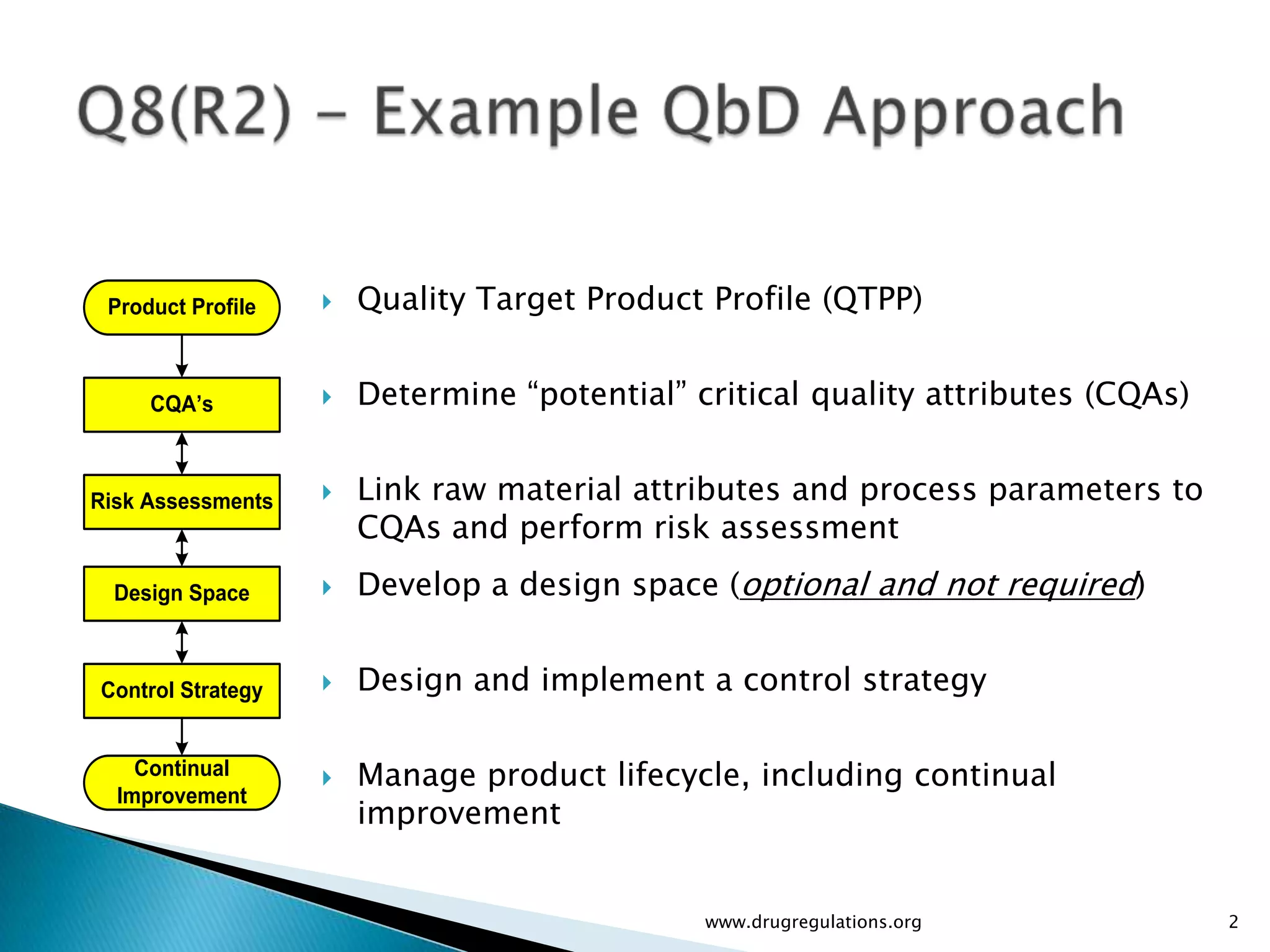
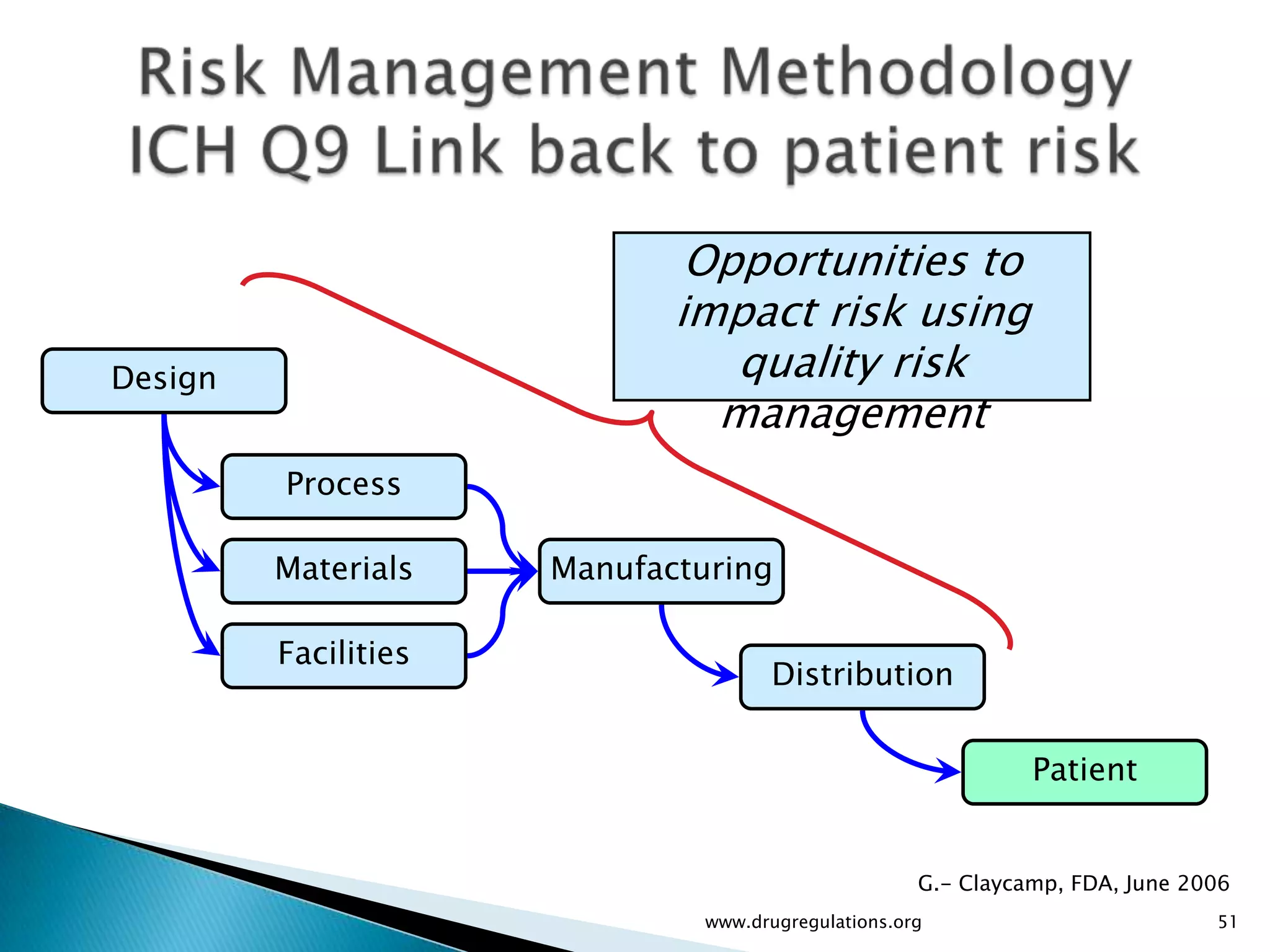
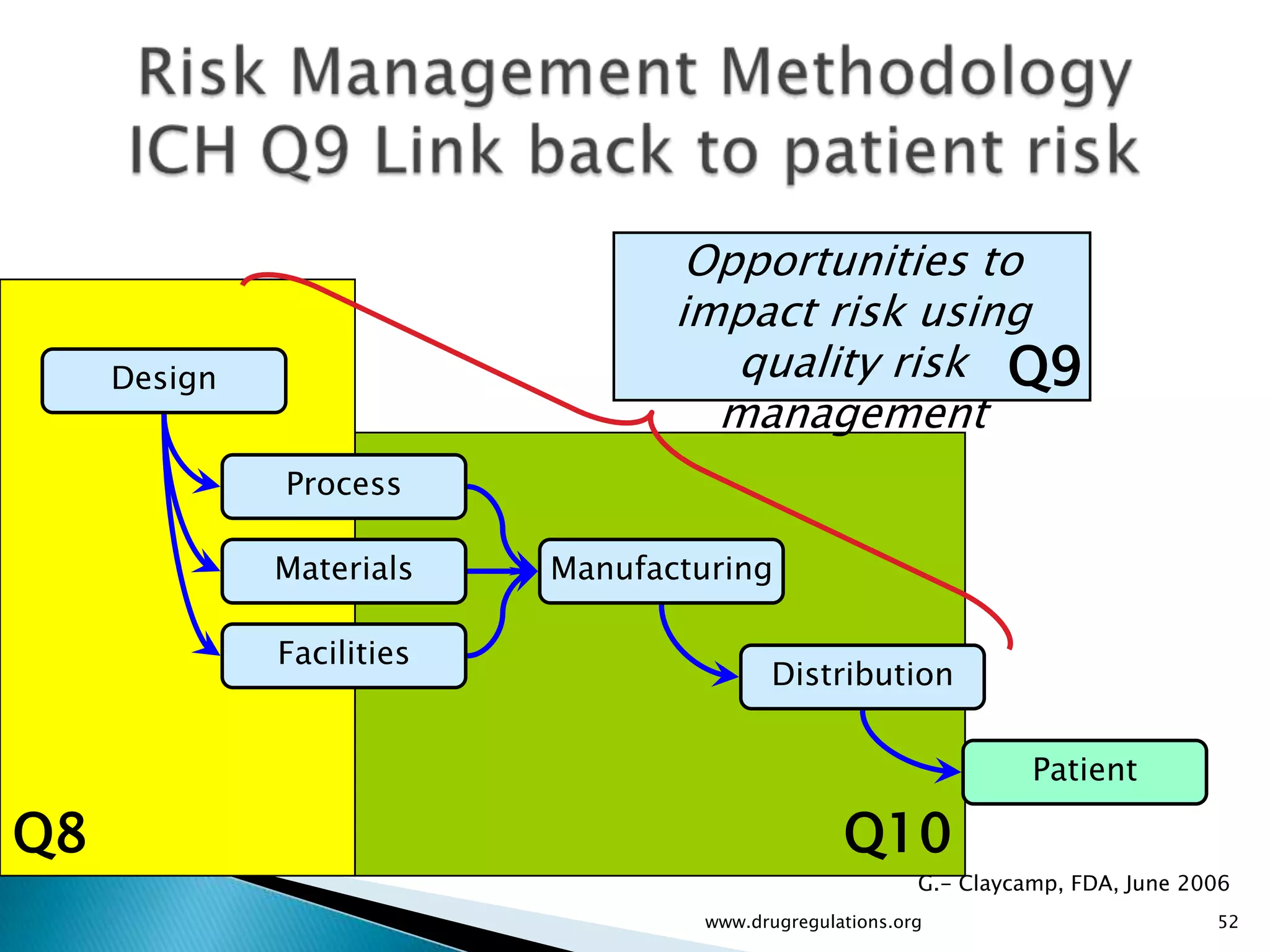
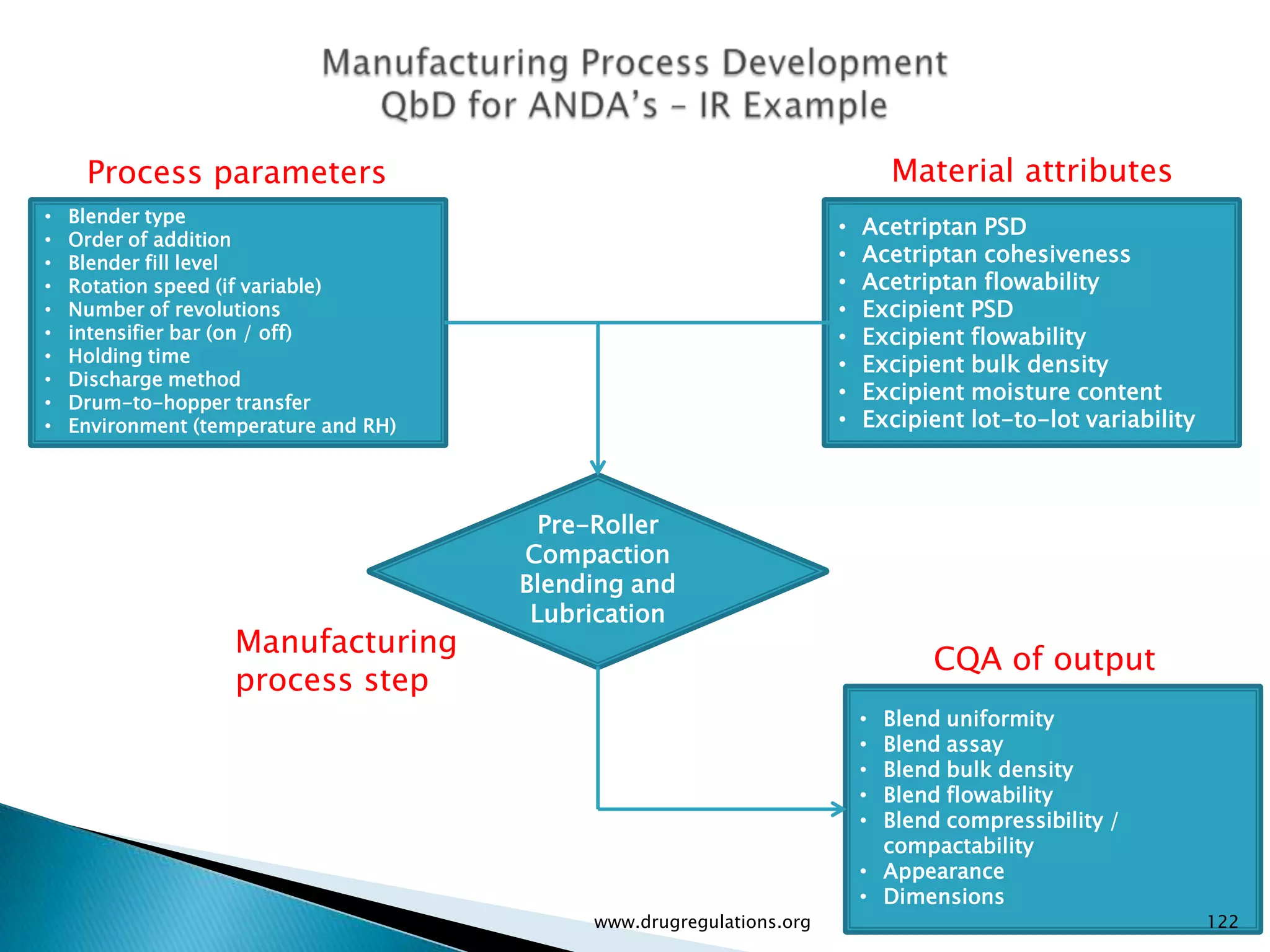
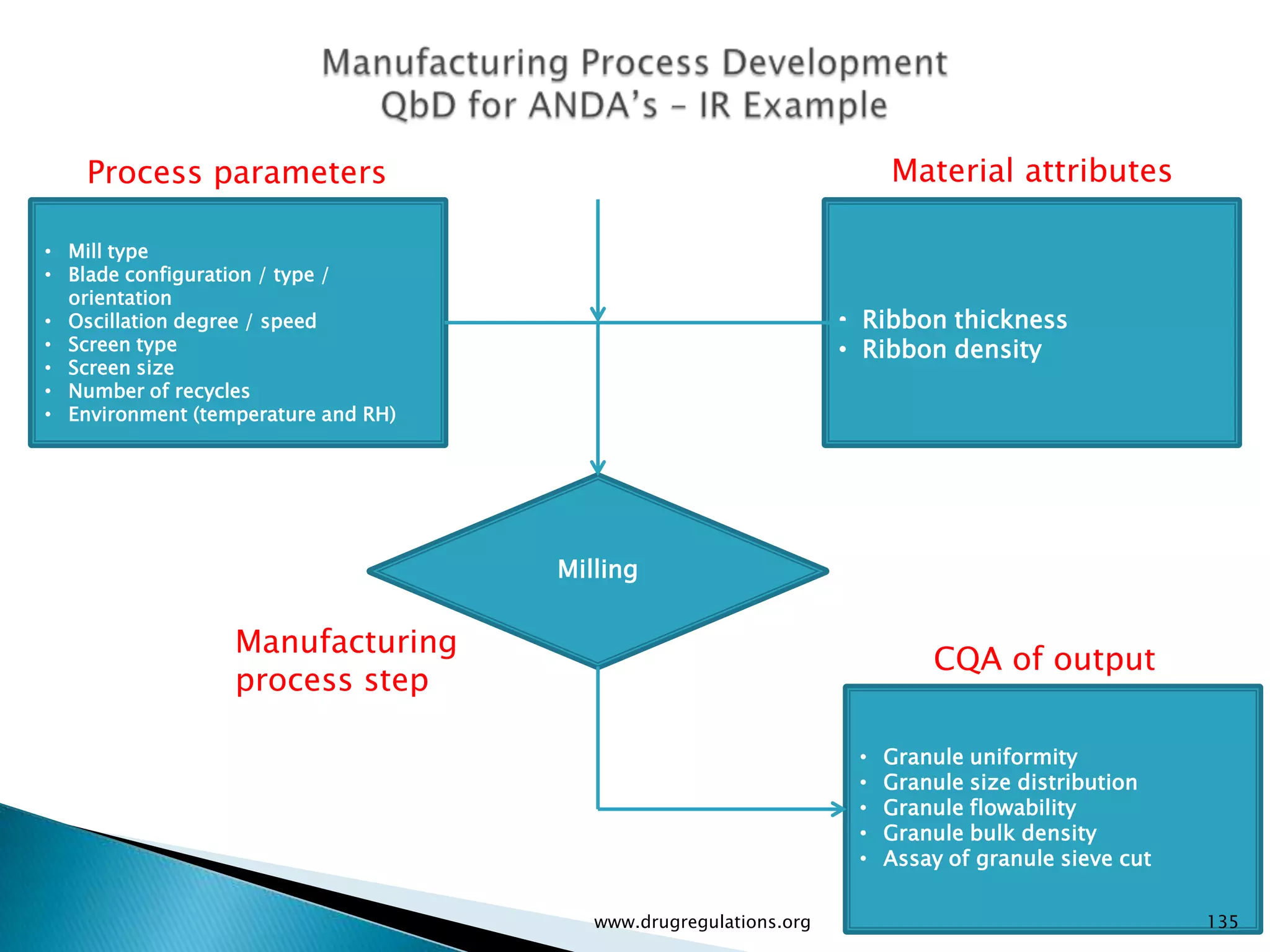
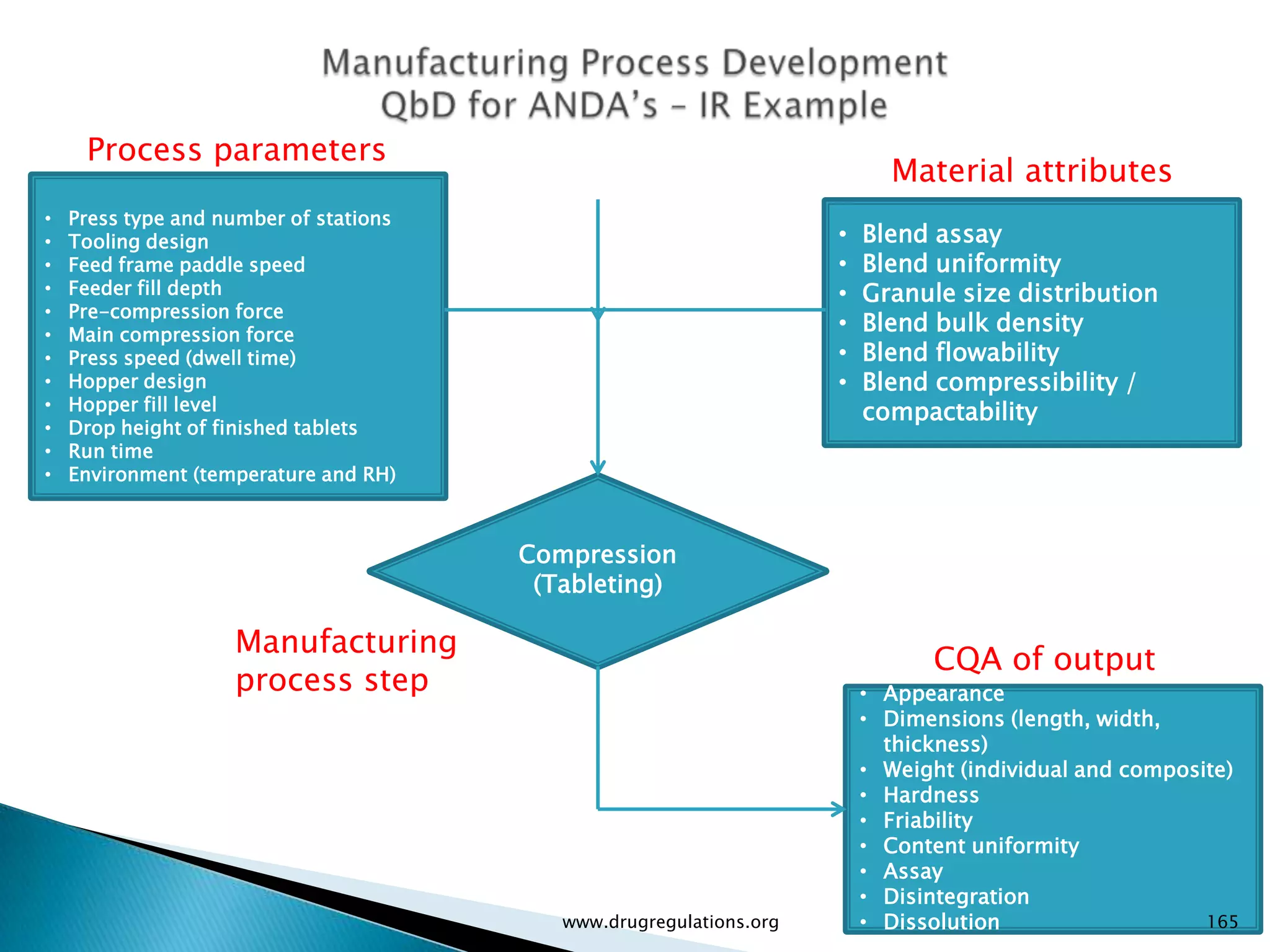
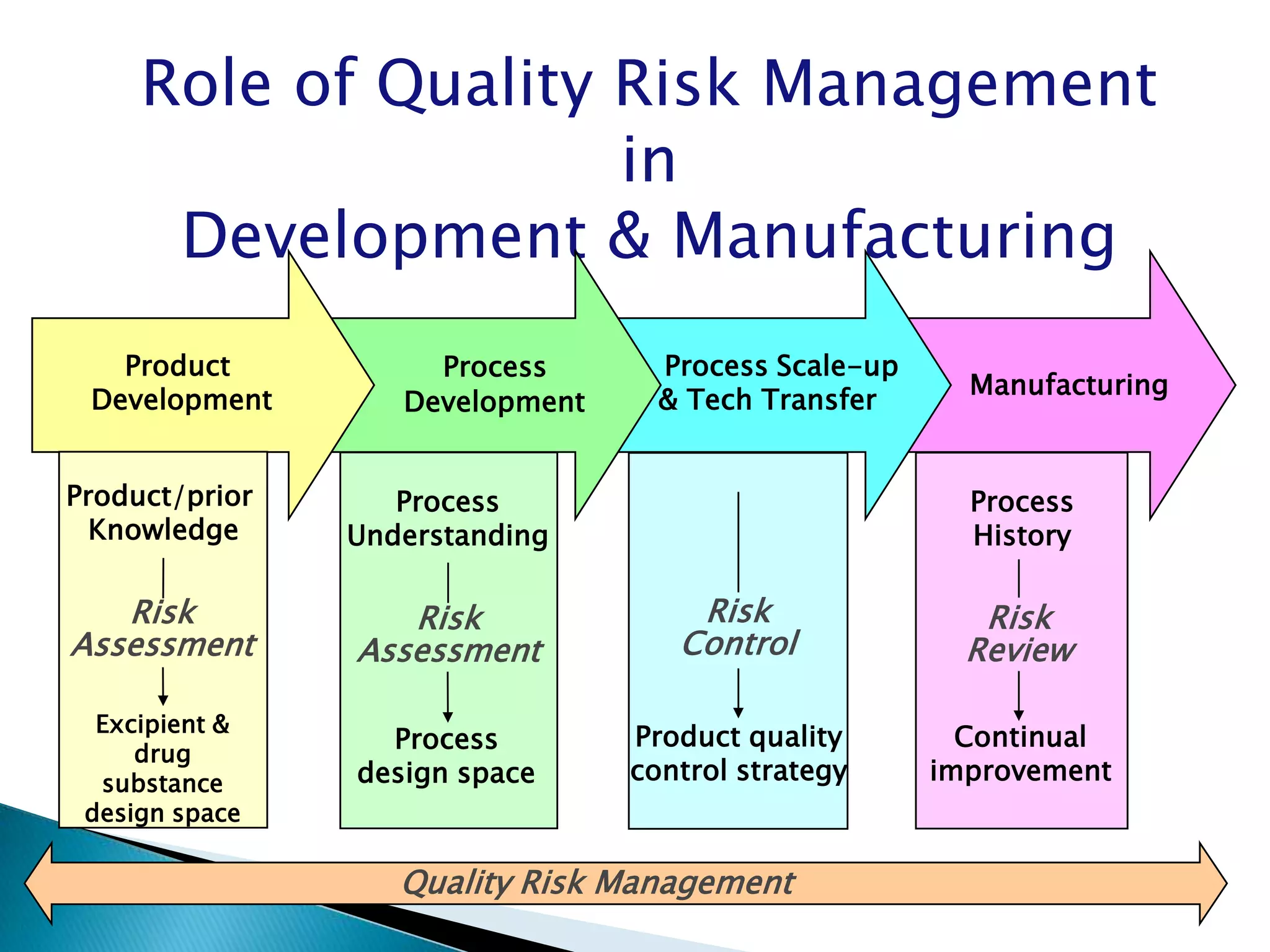
Overview of Risk Assessment linking material attributes and process parameters to Critical Quality Attributes (CQAs) in pharmaceutical development.
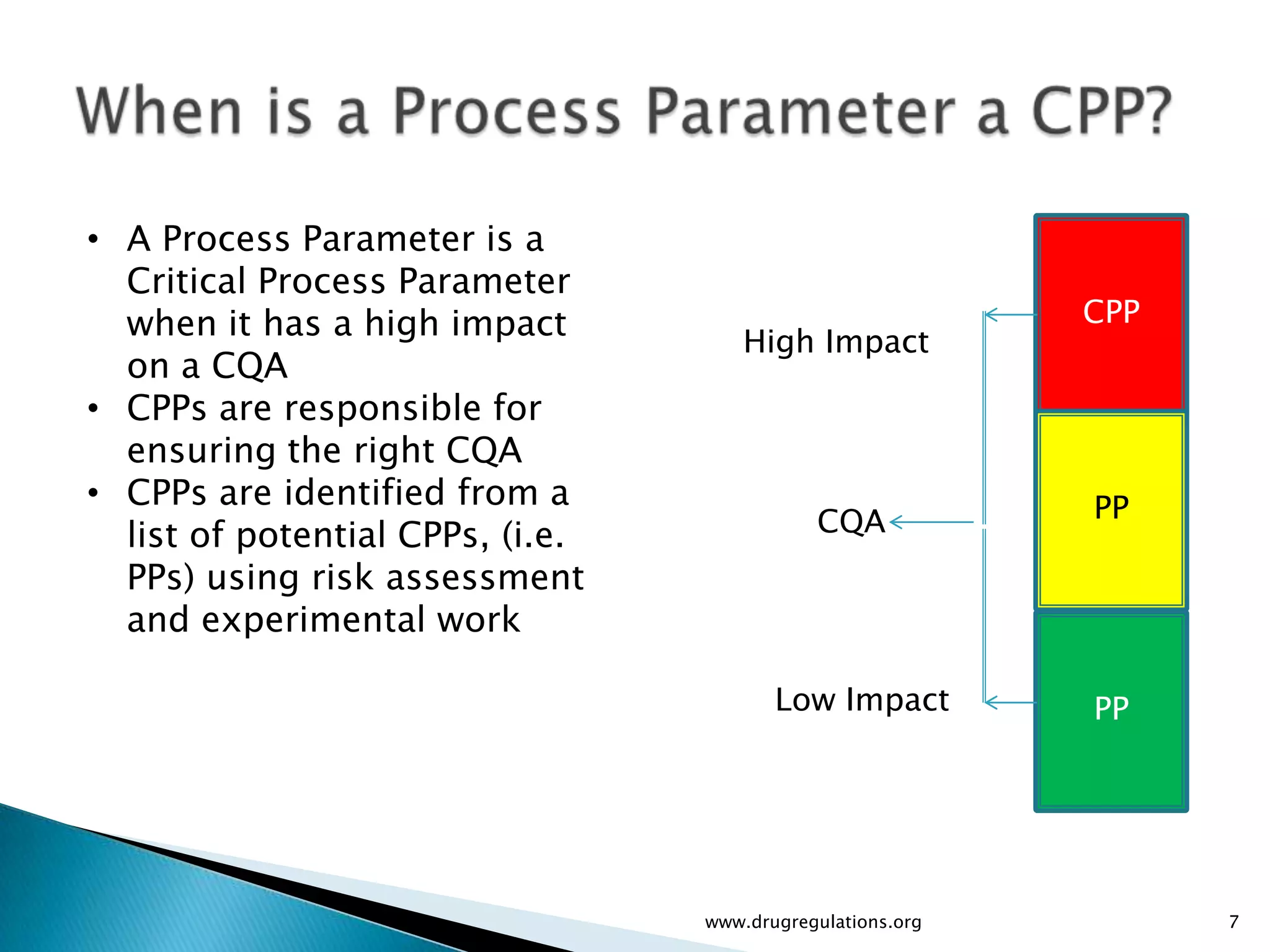
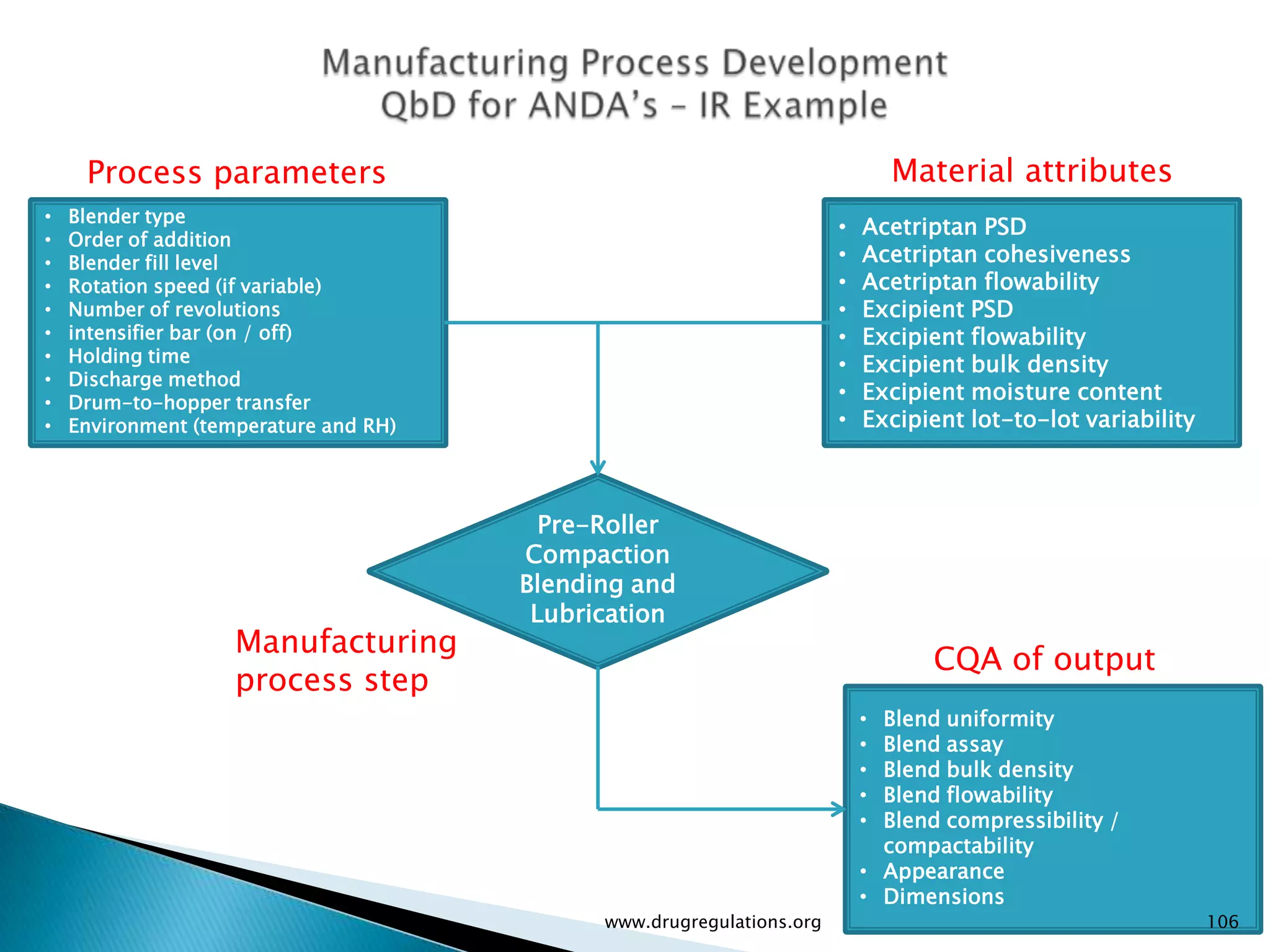
Definitions and examples of material attributes and process parameters critical for ensuring drug product quality.
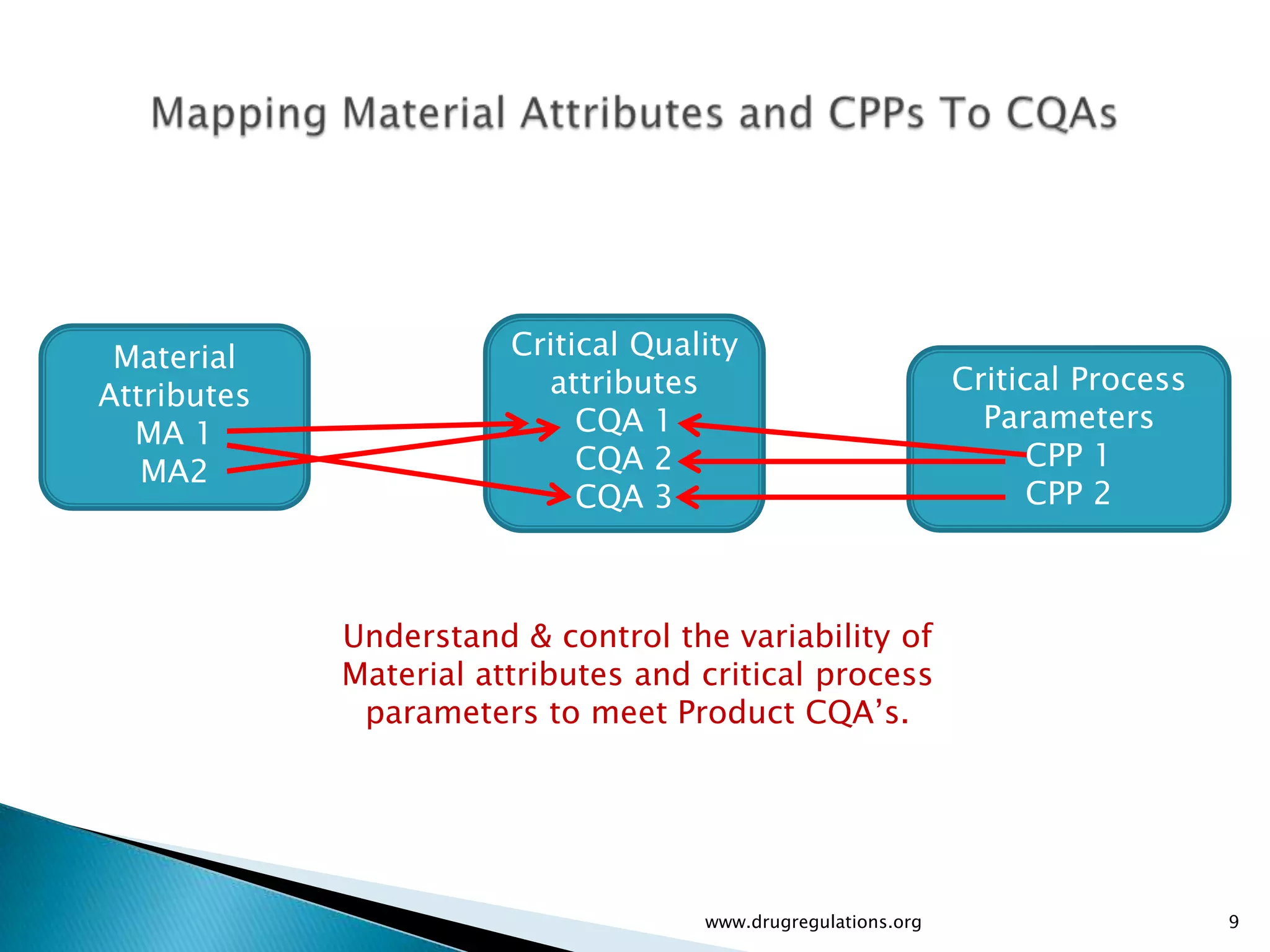
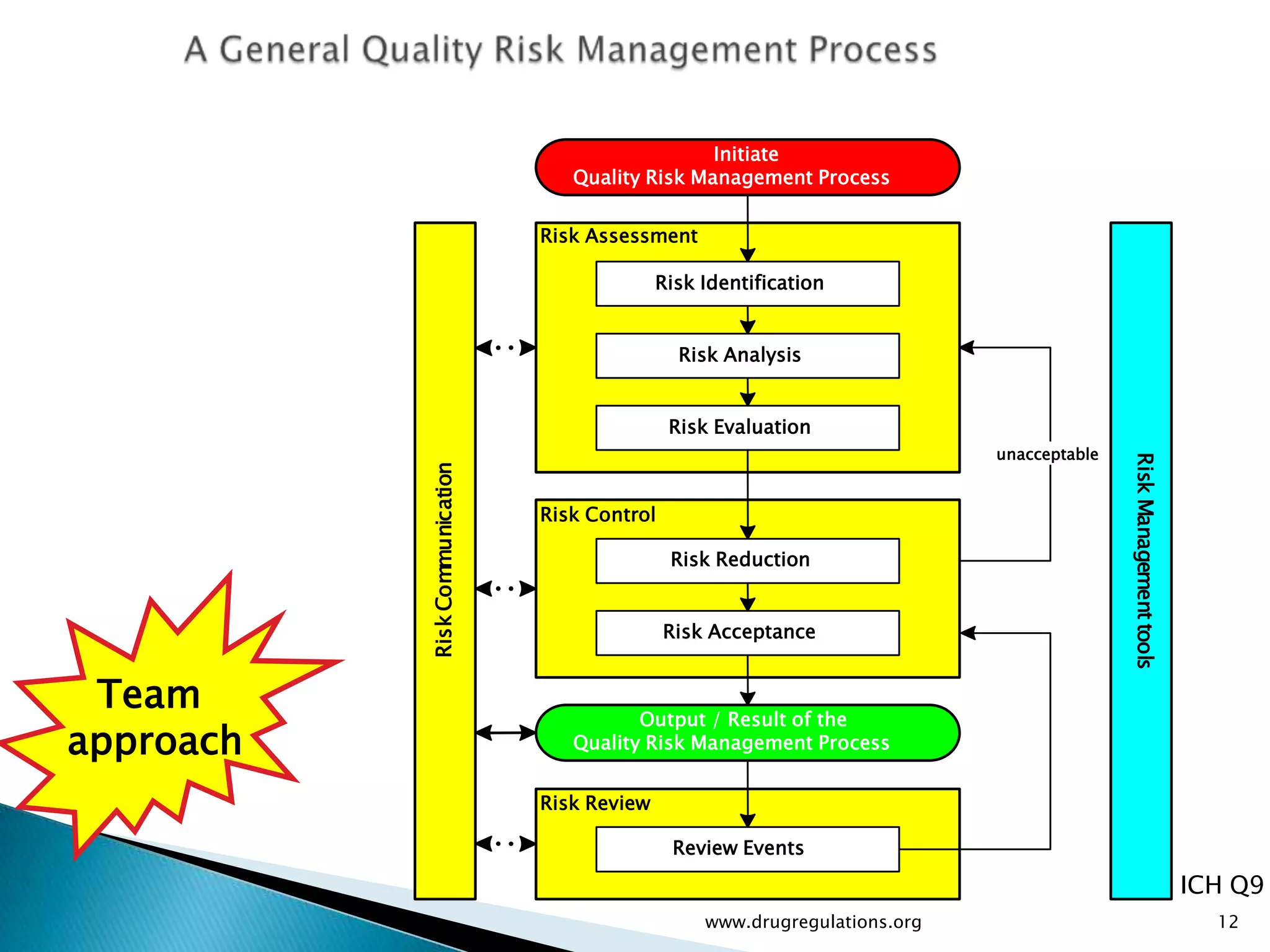
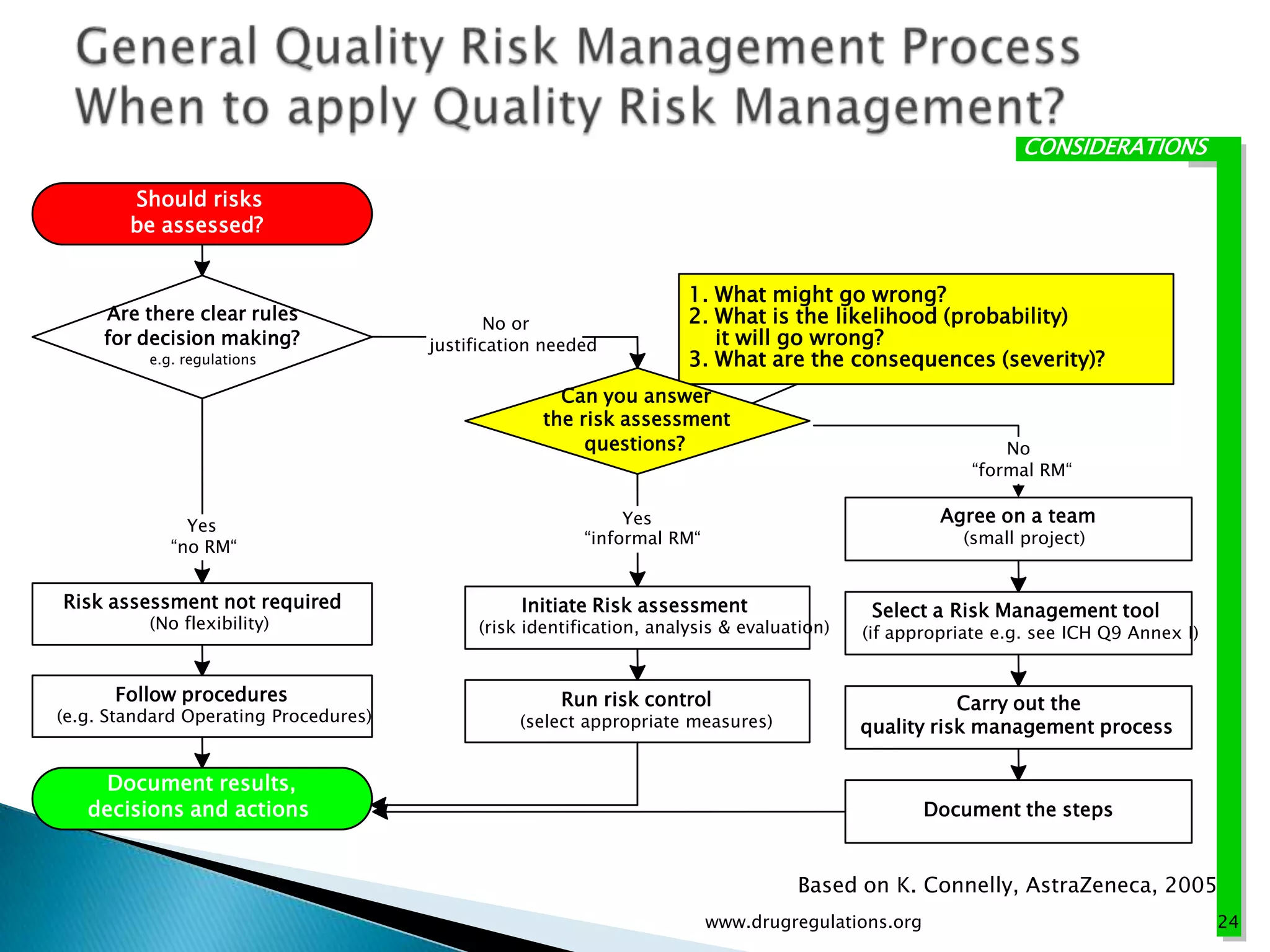
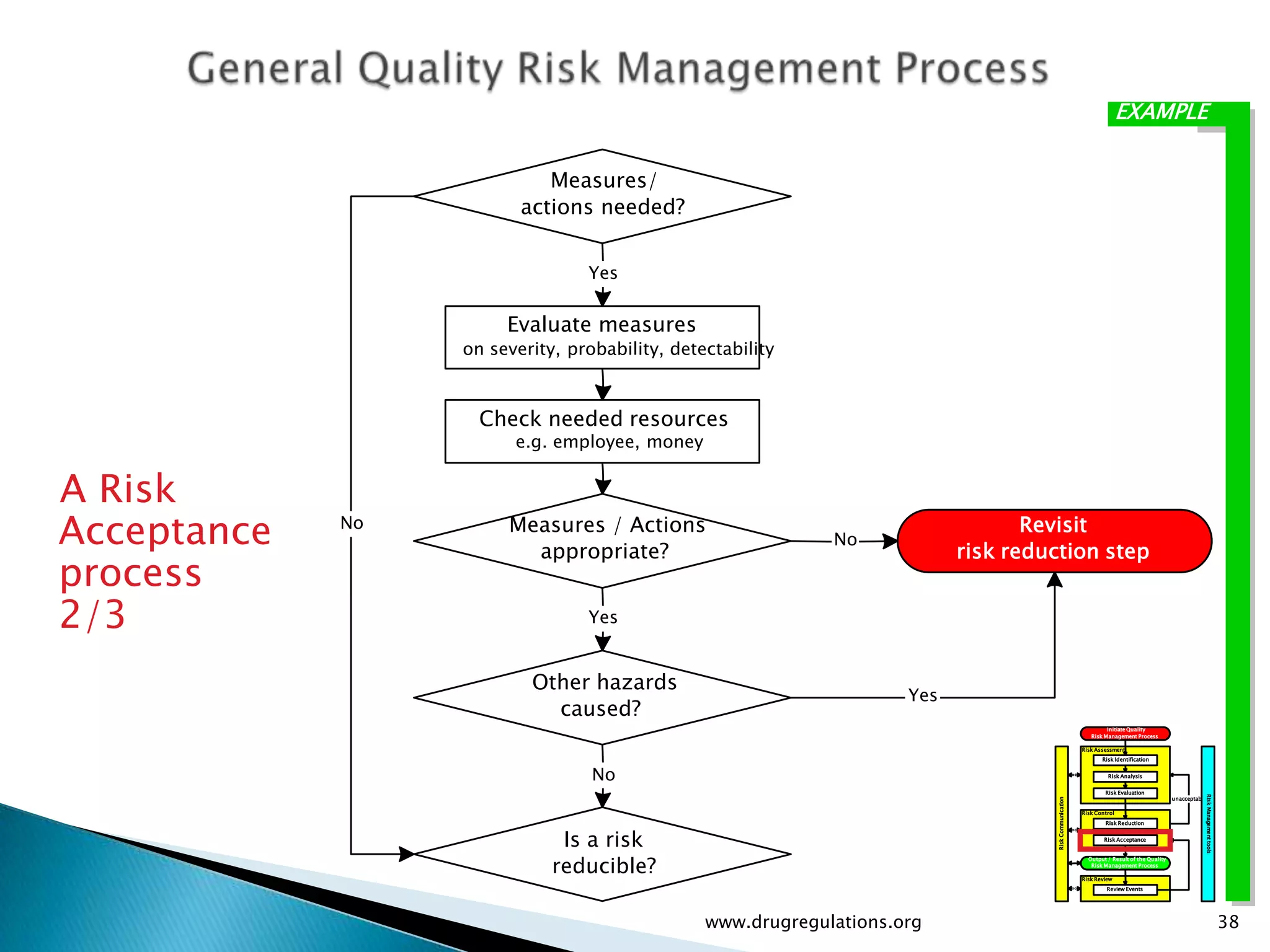
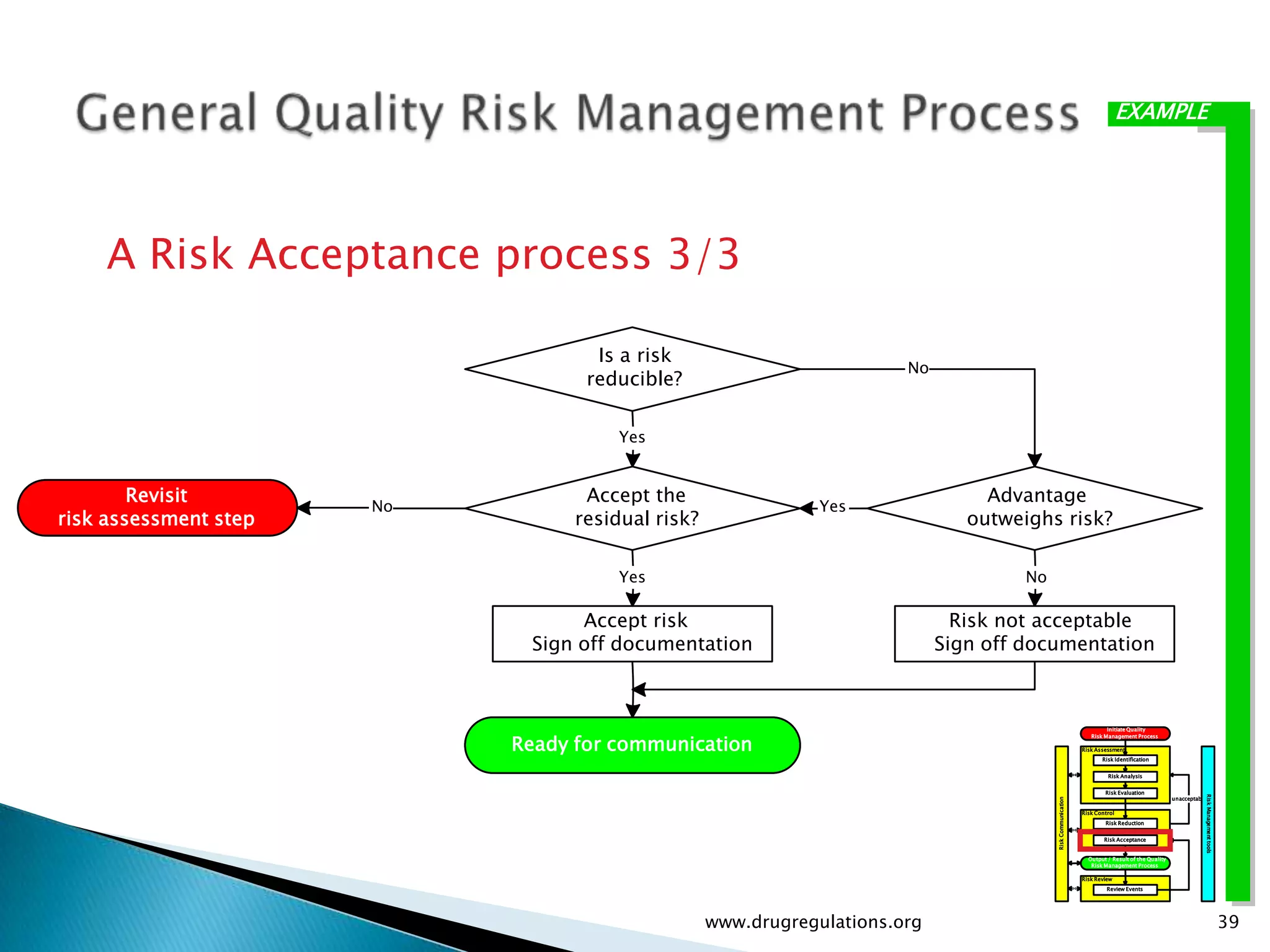
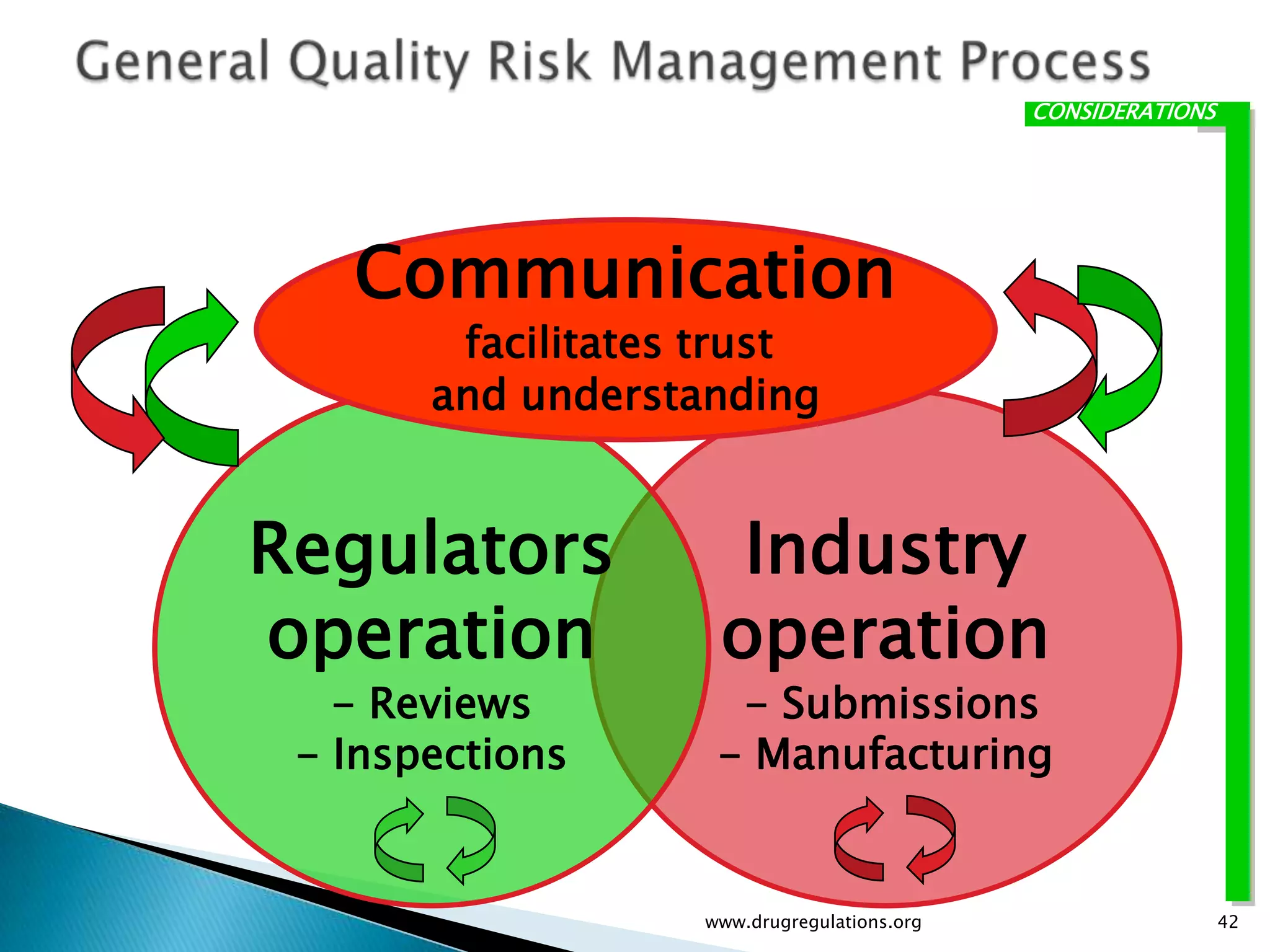
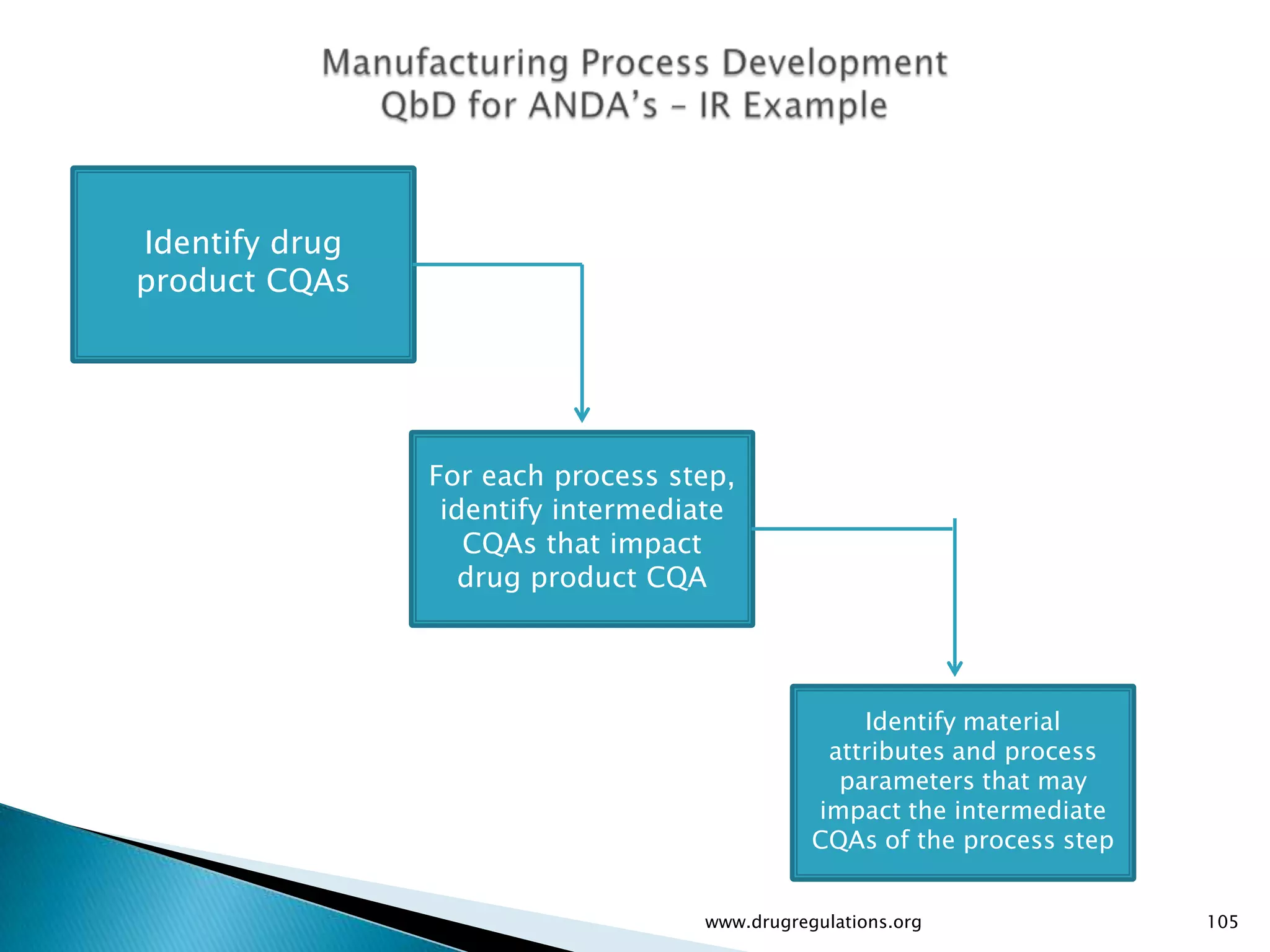
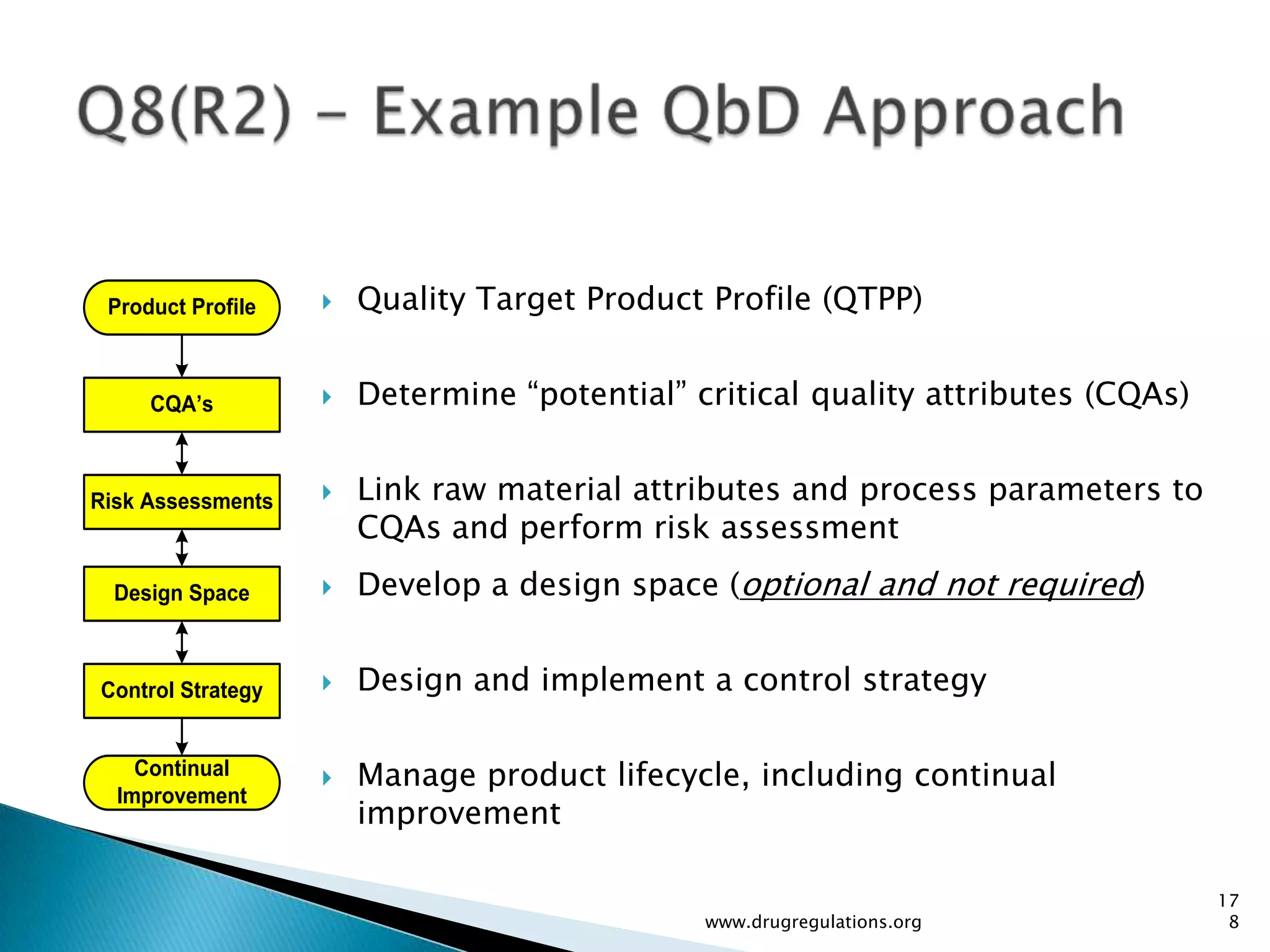
Discussion on linking Material Attributes (MAs) and Critical Process Parameters (CPPs) to ensure product CQAs are achieved.Introduction to the systematic Quality Risk Management process, including risk assessment, communication, and control strategies associated with CQAs.
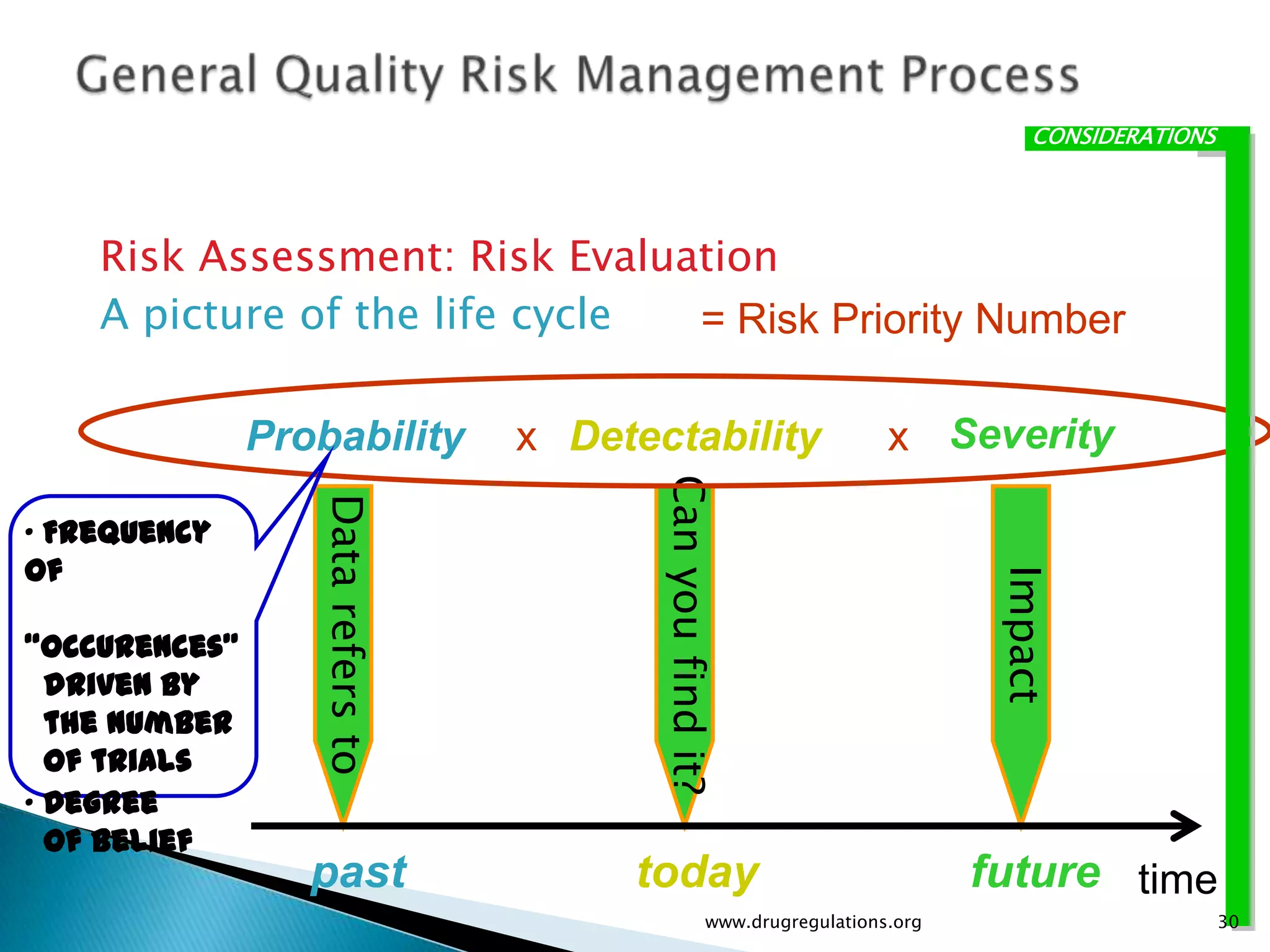
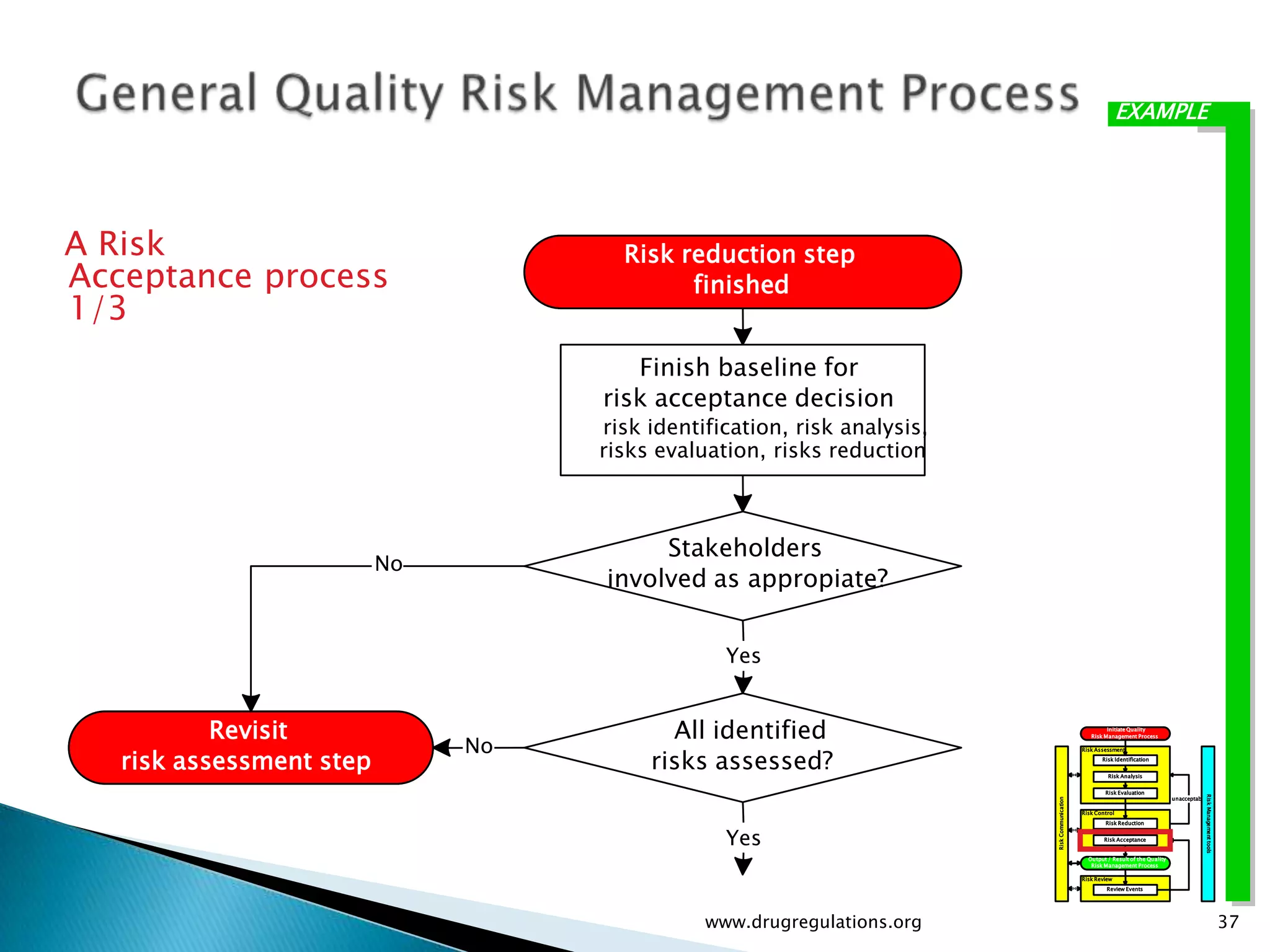
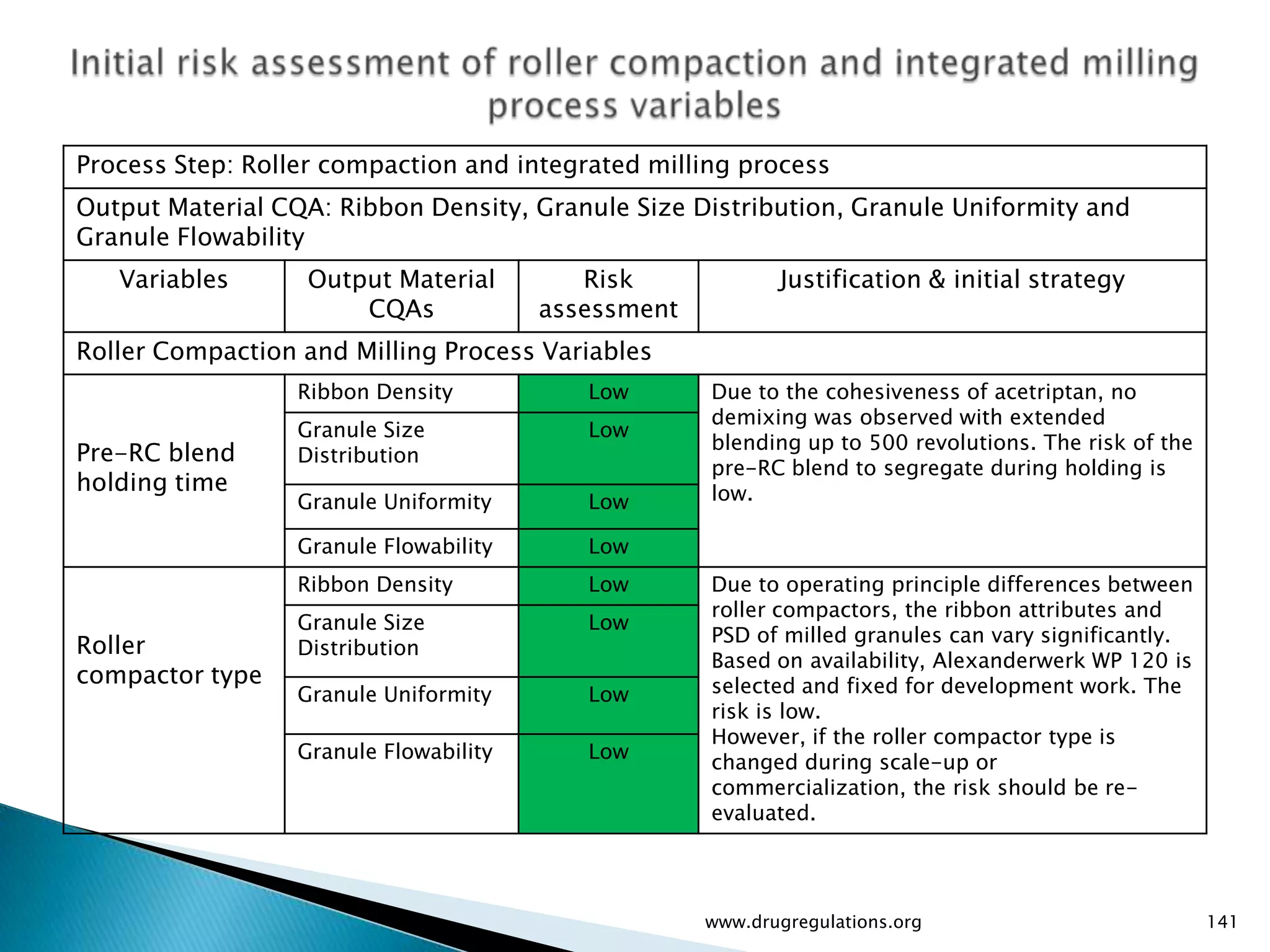
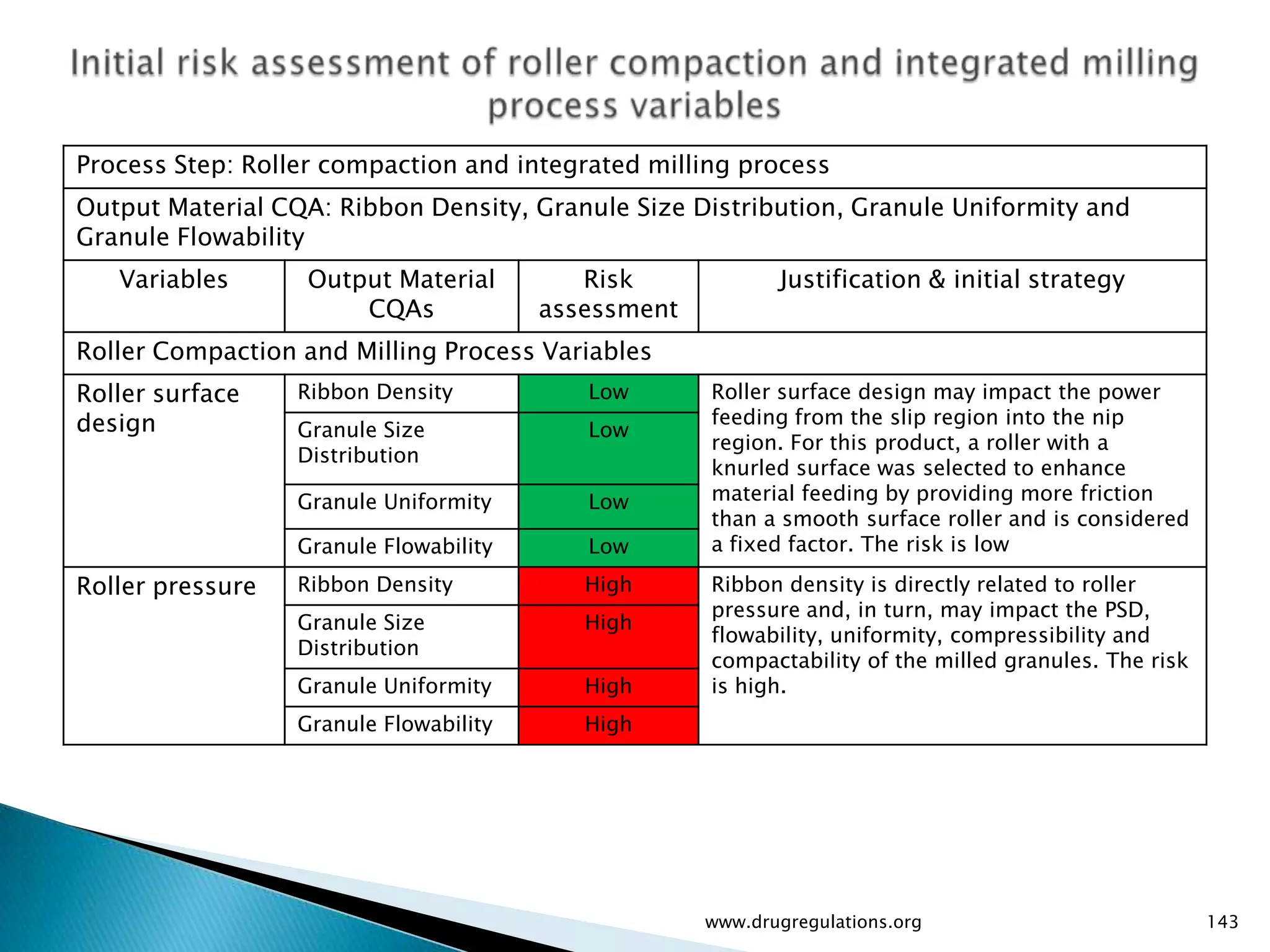
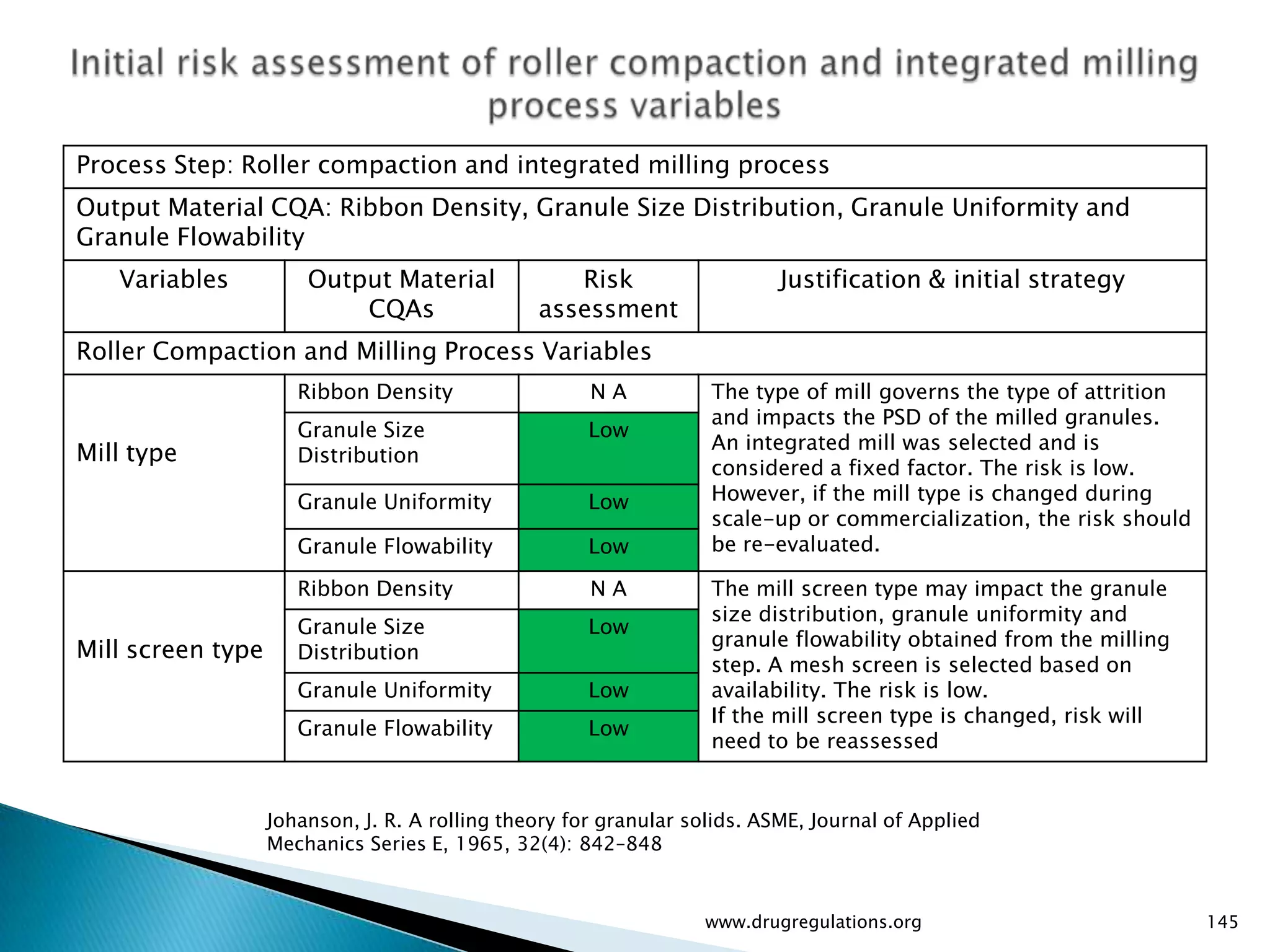
Key definitions in risk analysis including risk communication, analysis, evaluation, and reduction related to pharmaceutical processes.
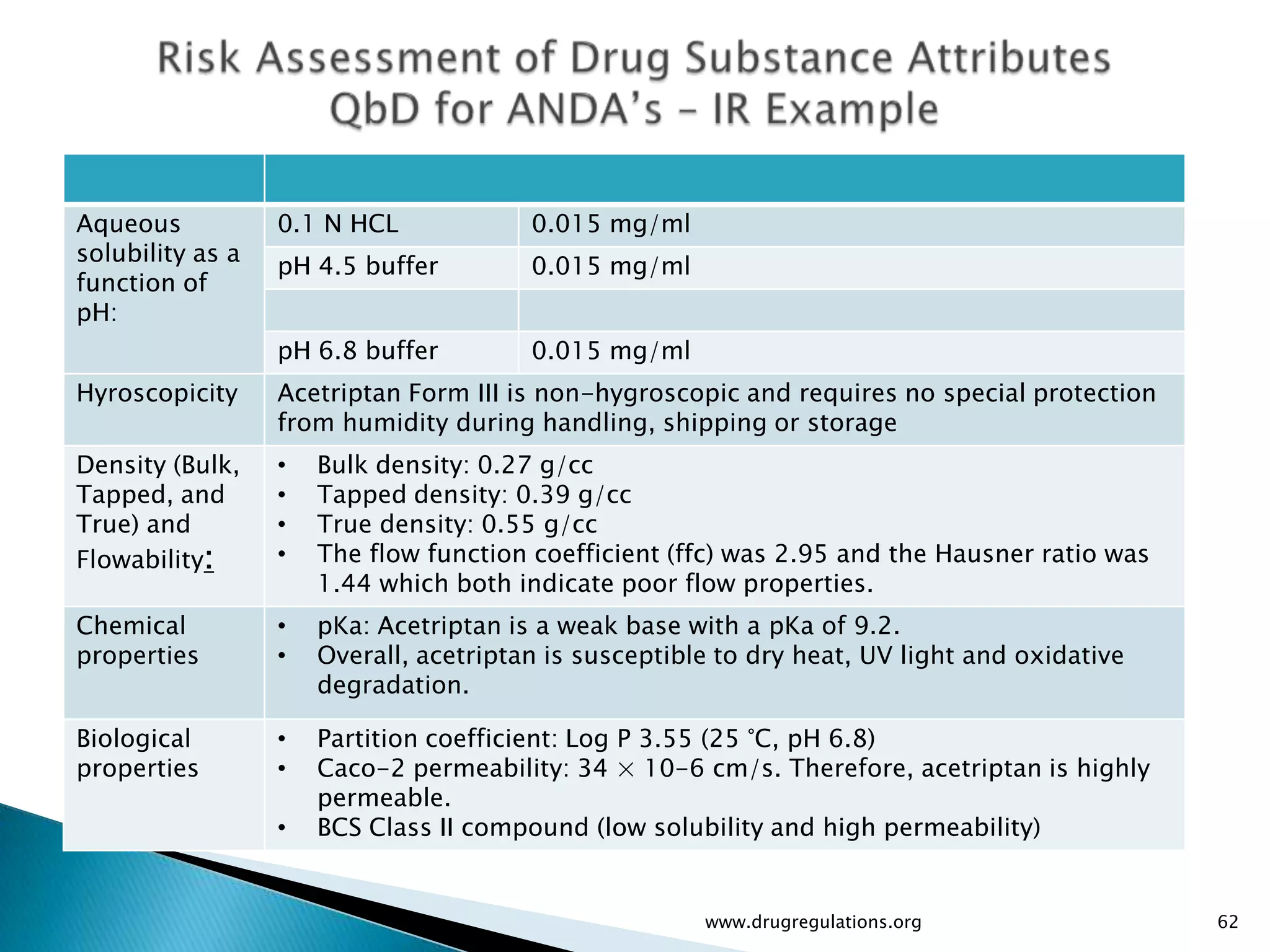
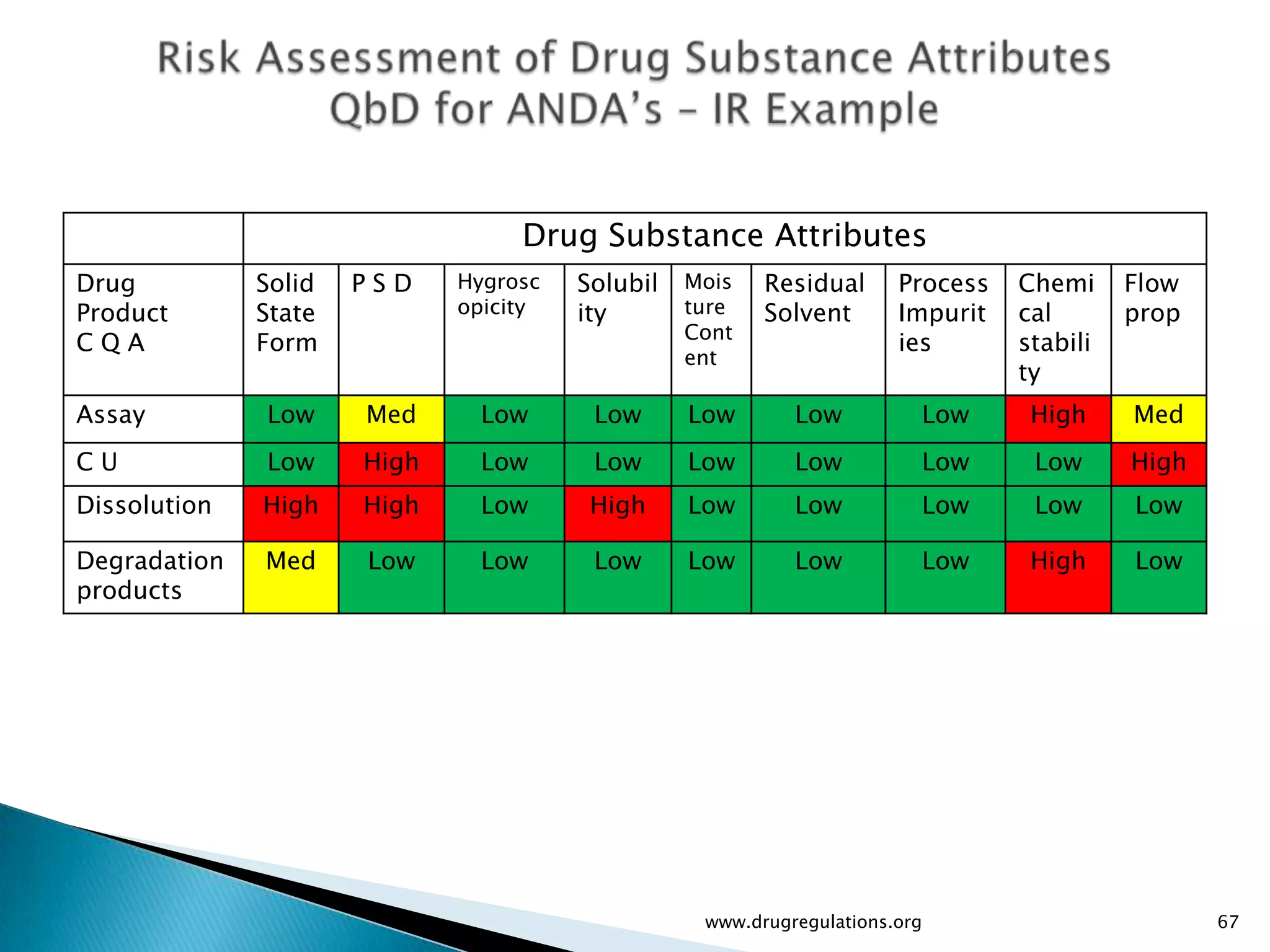
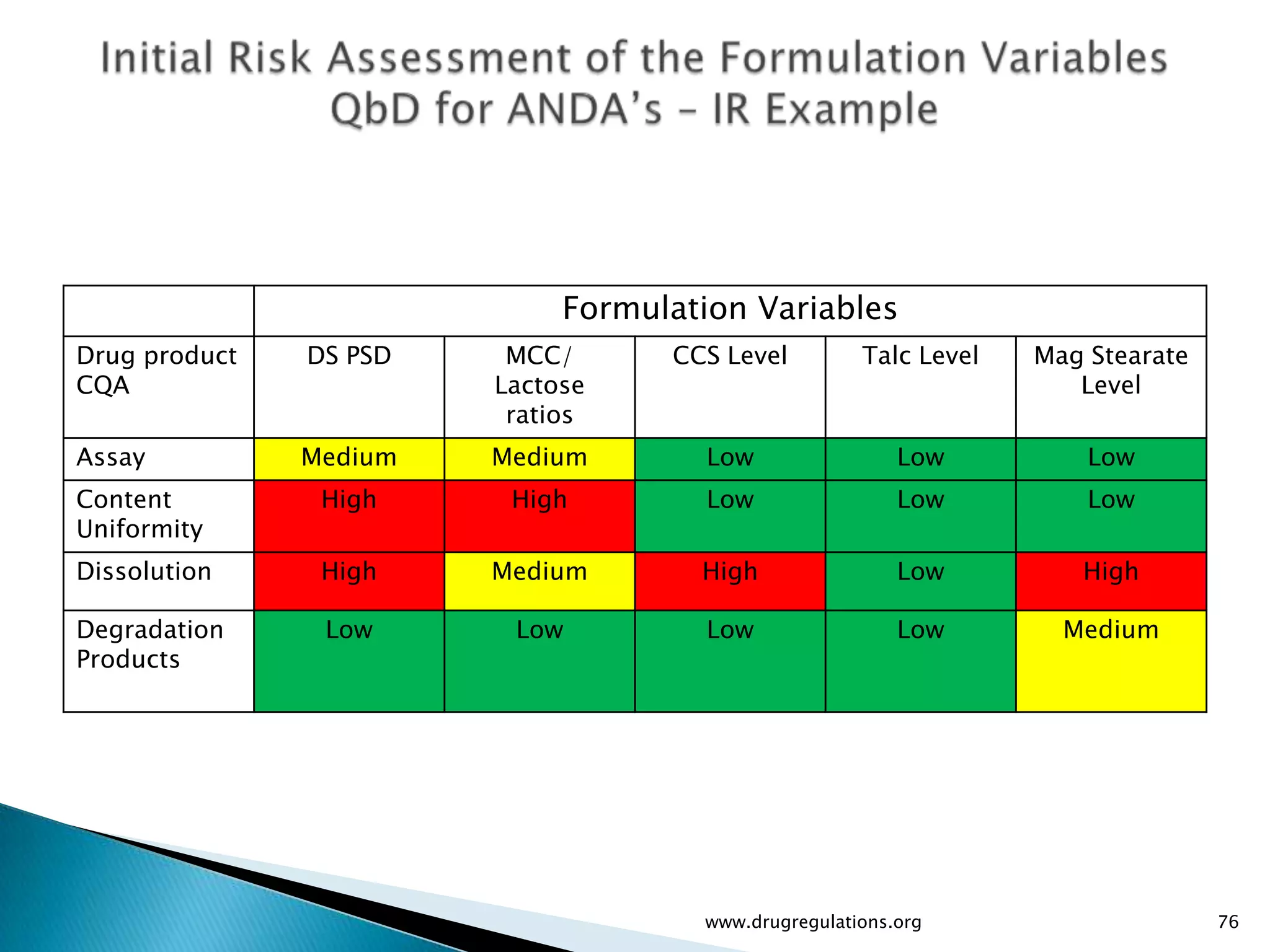
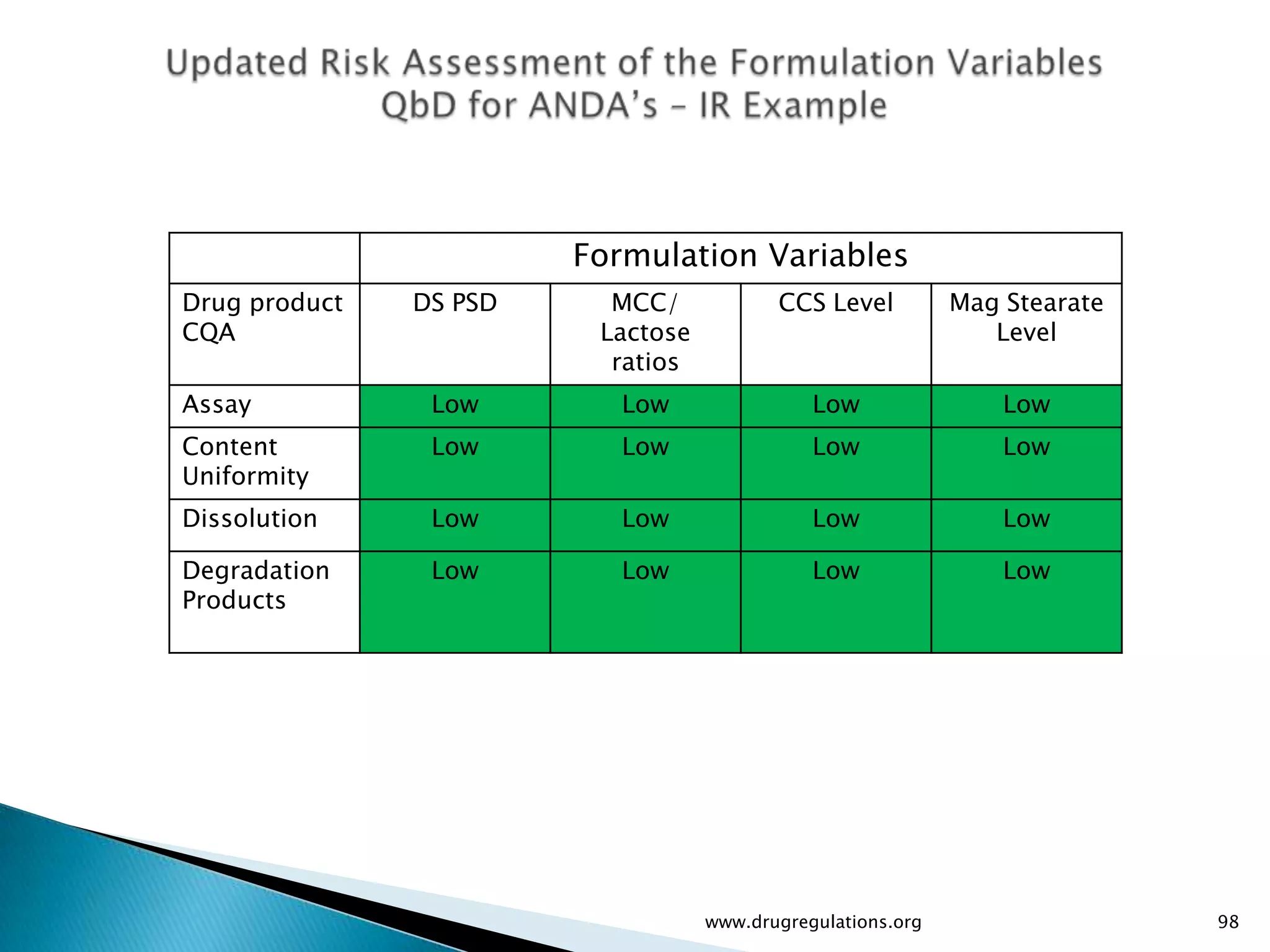
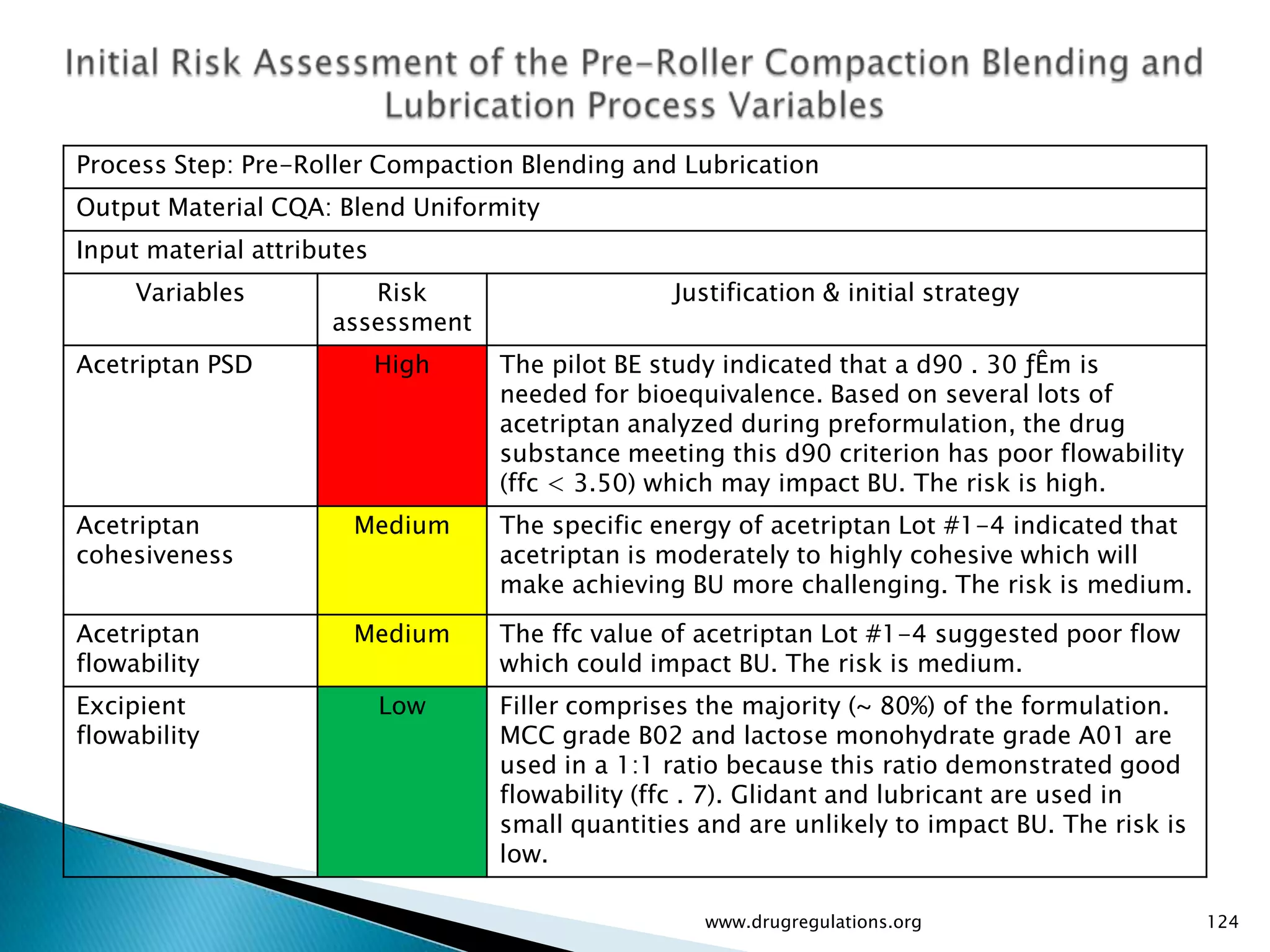
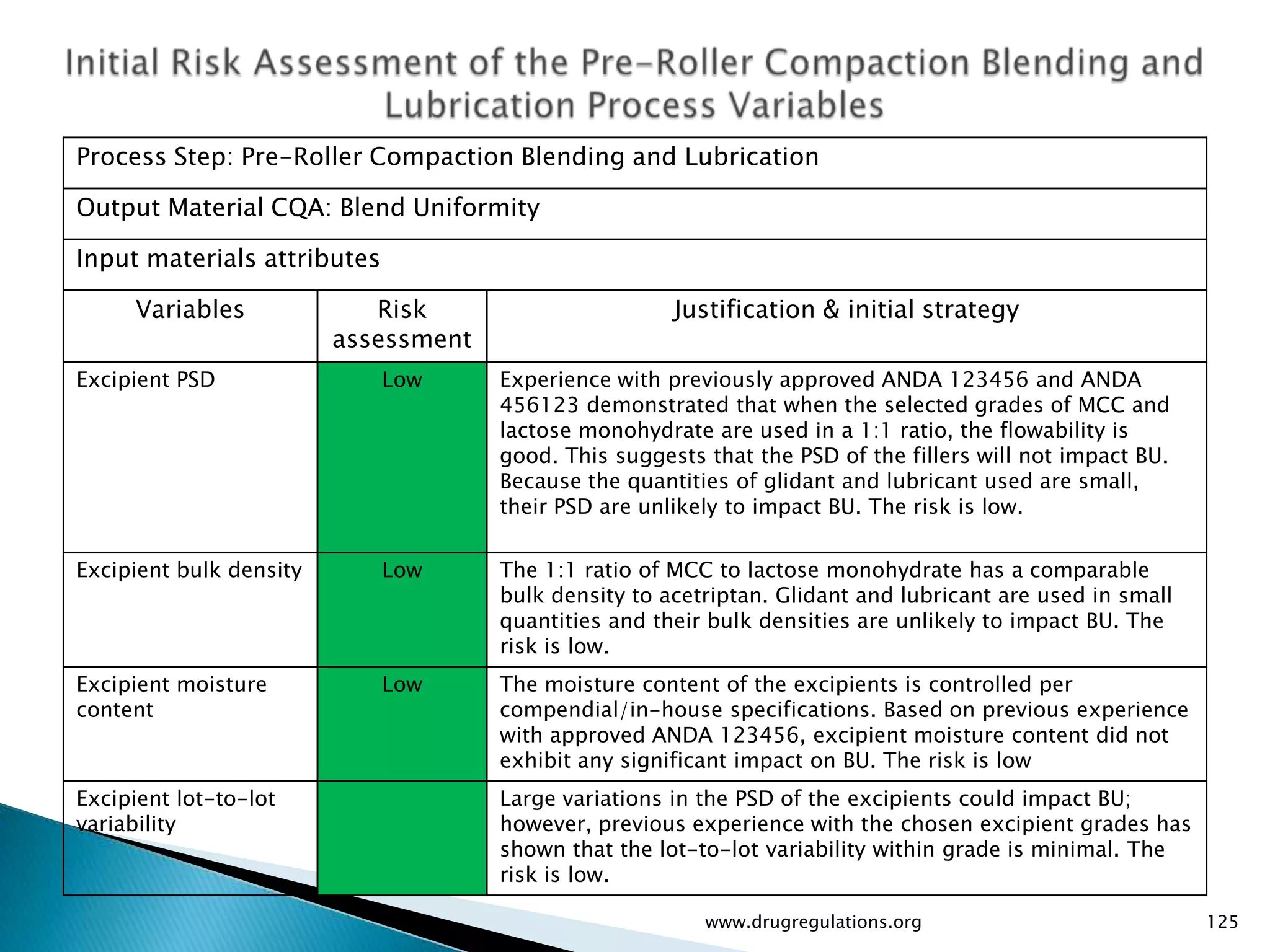
Assessment of drug substance attributes regarding their impact on the formulation and processes affecting CQAs.
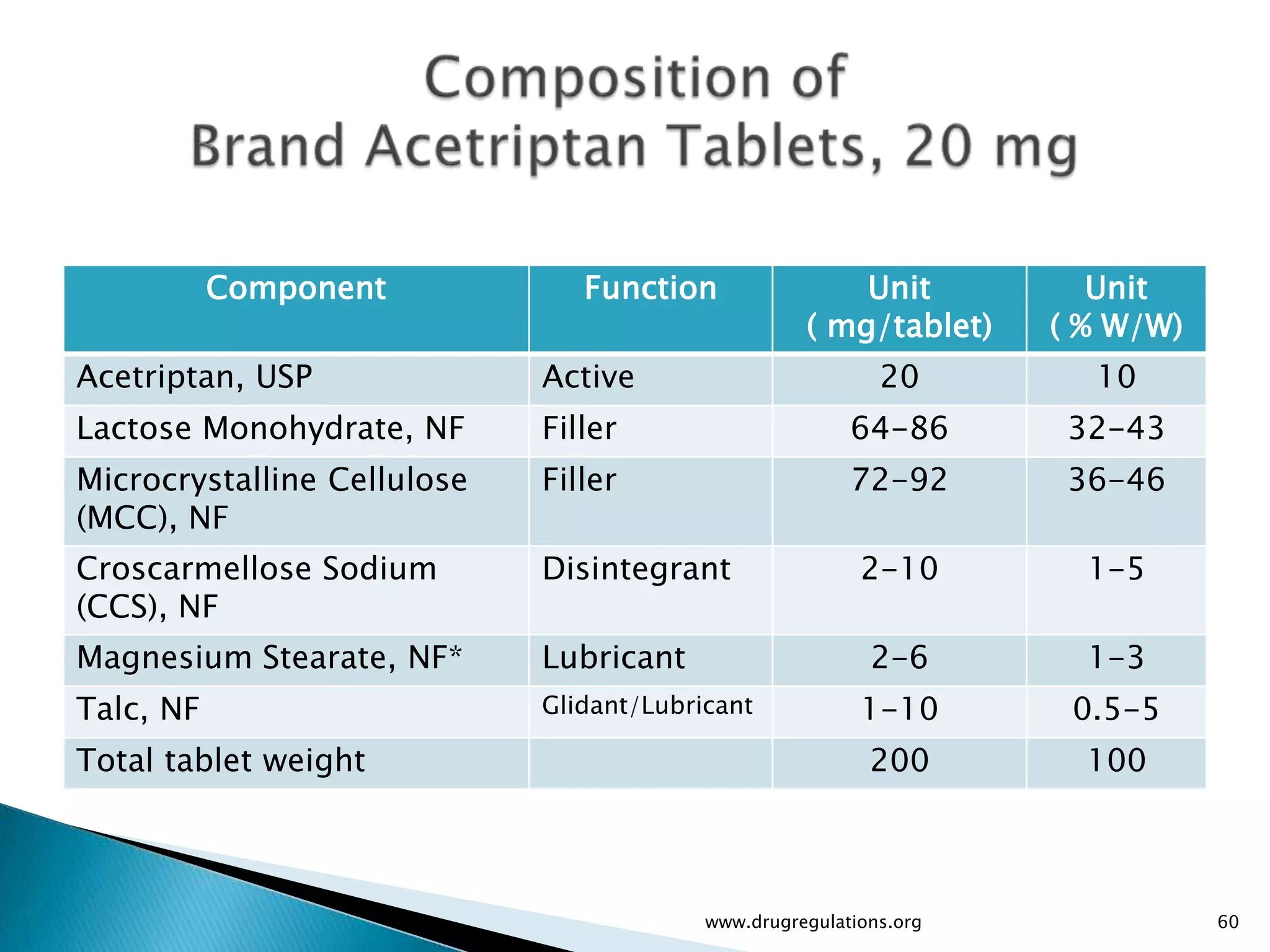
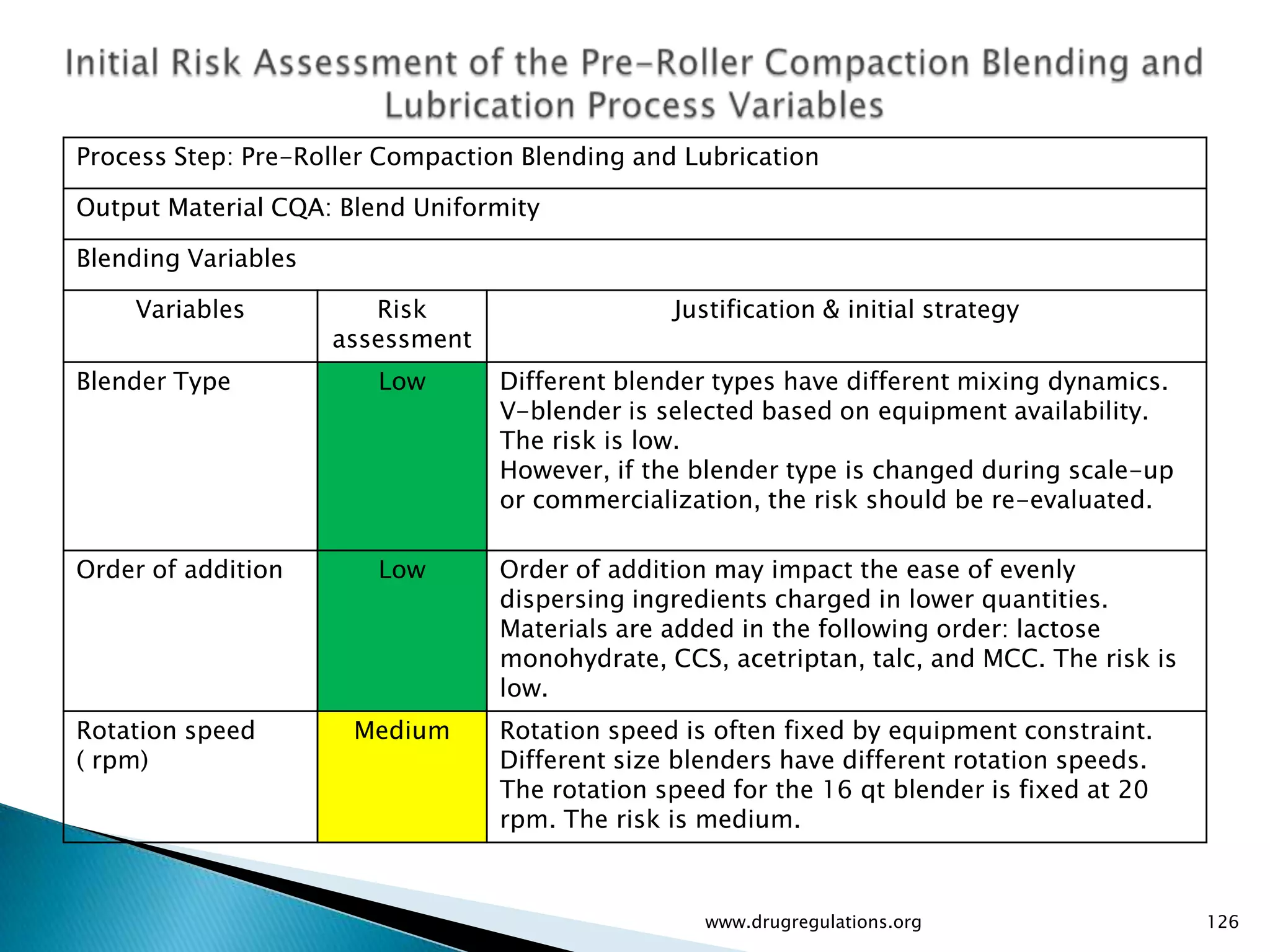
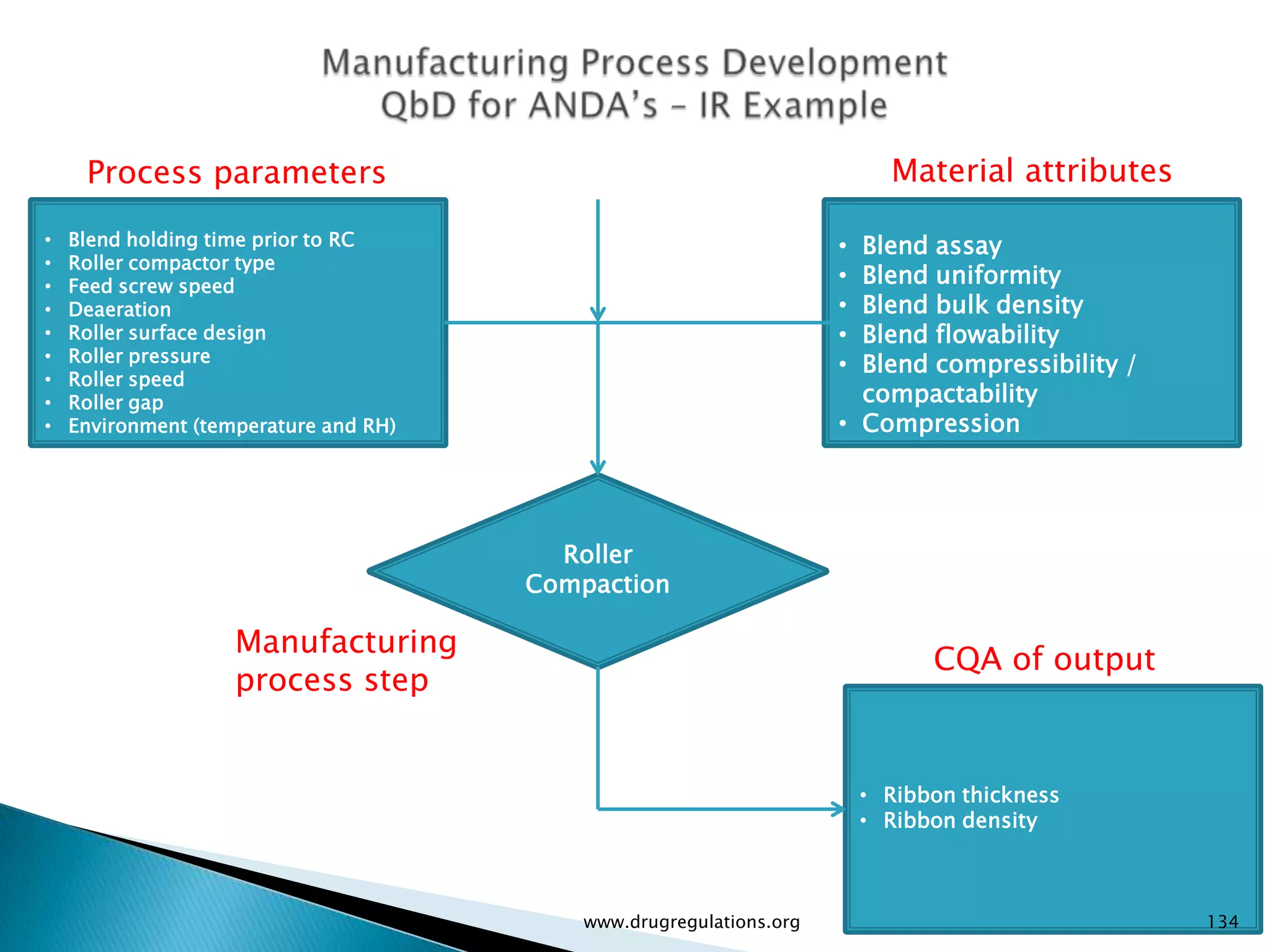
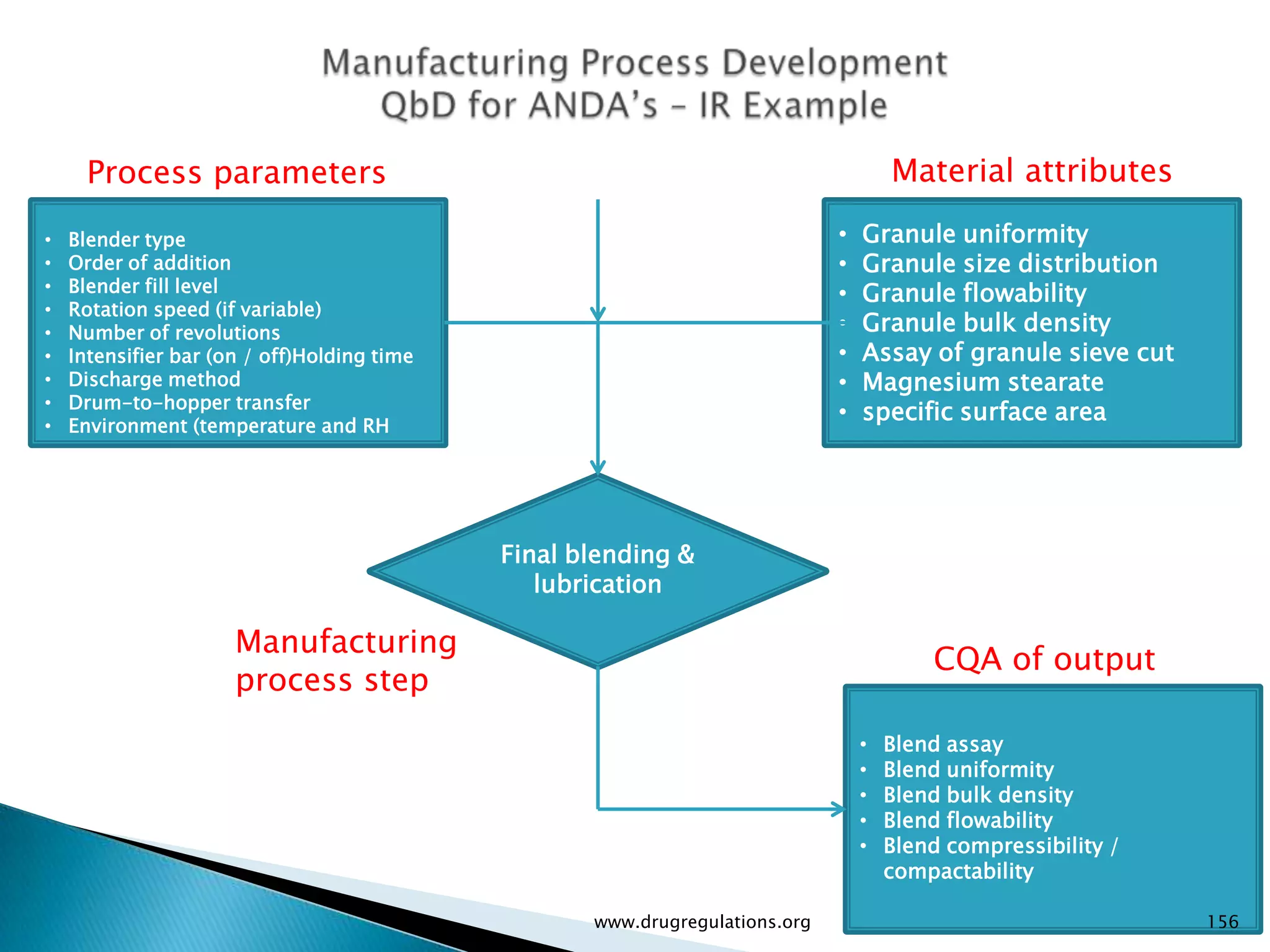
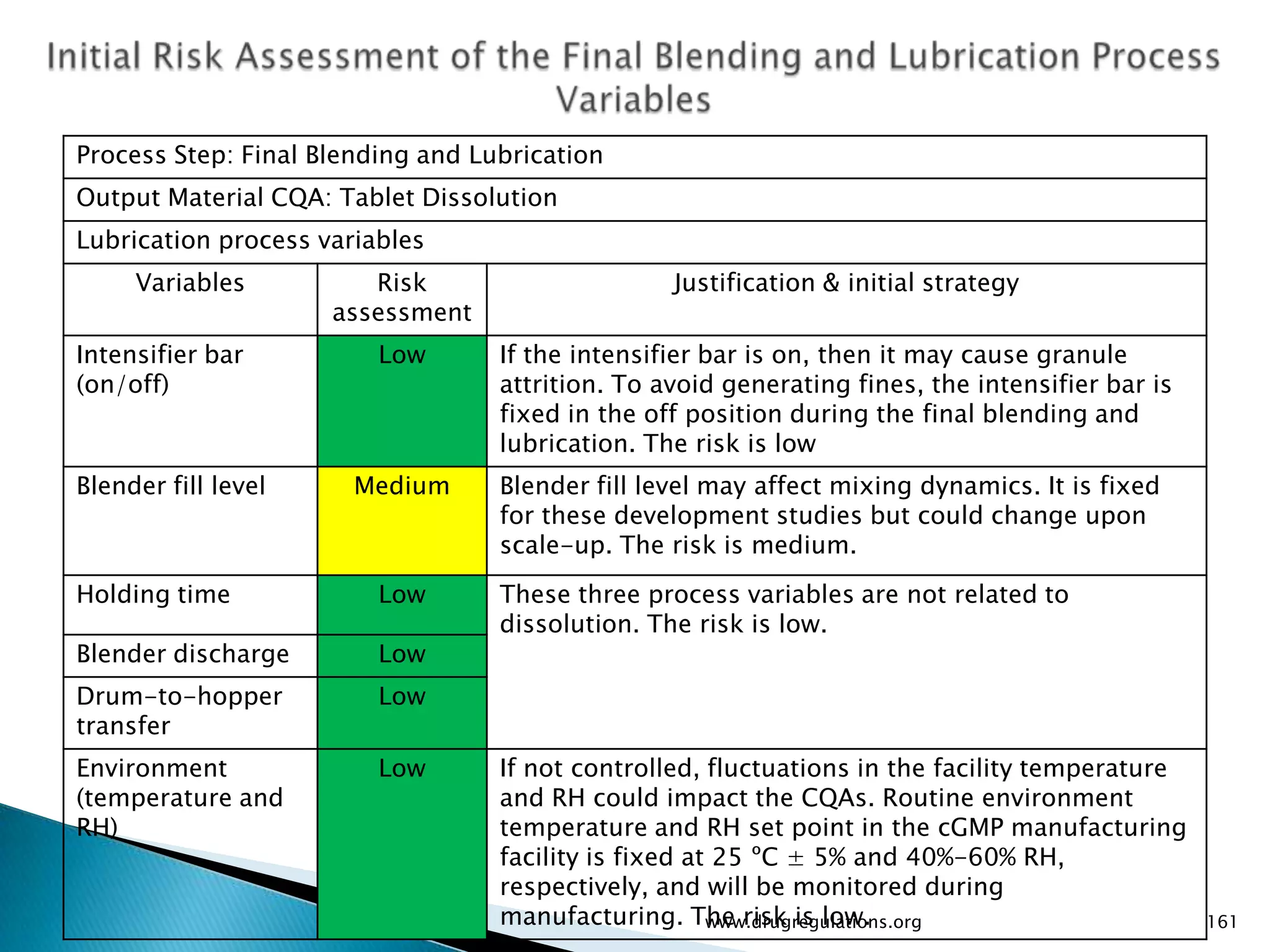
Details on formulation development studies focusing on variables impacting CQAs and process parameters during manufacturing.
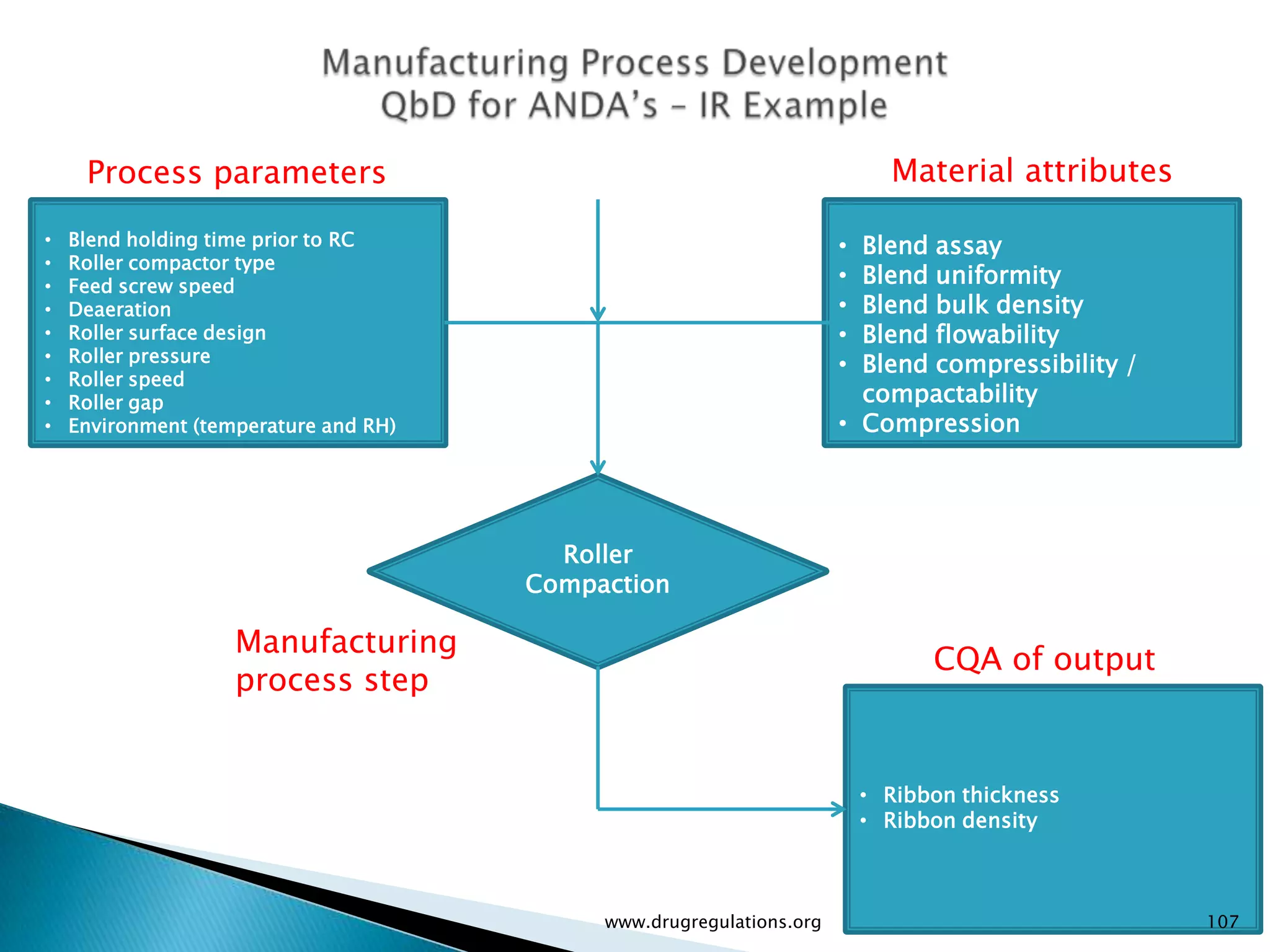
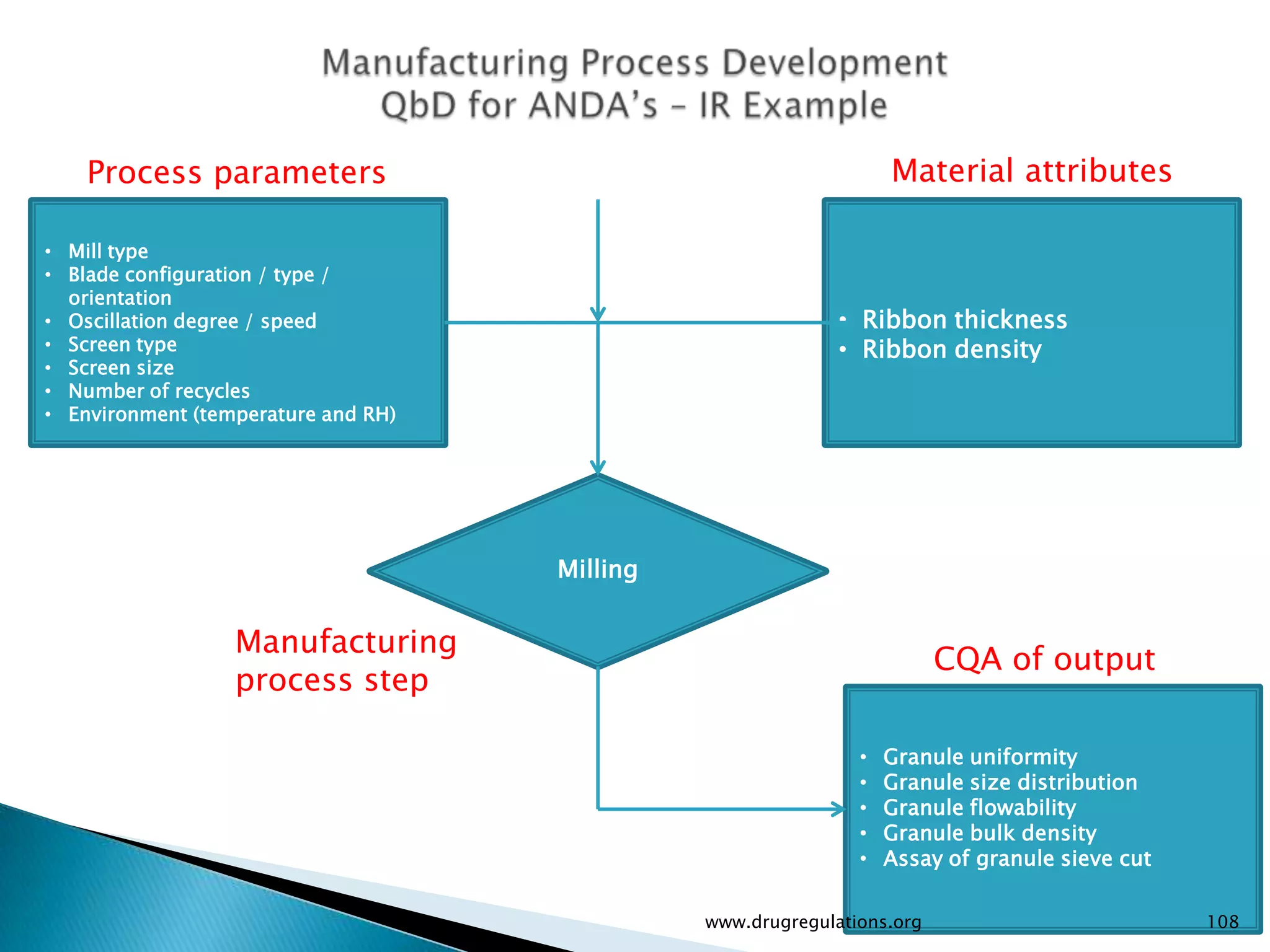
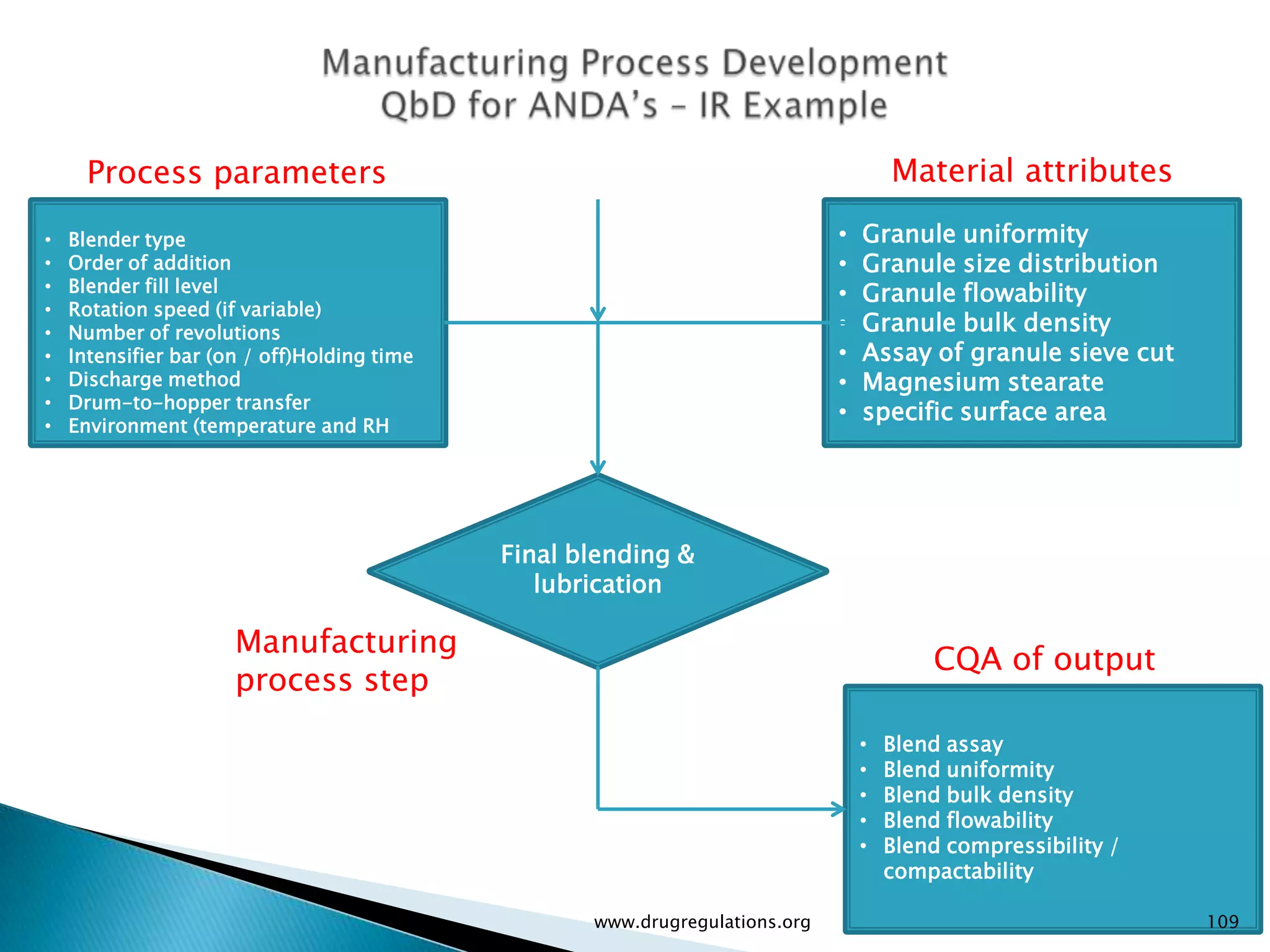
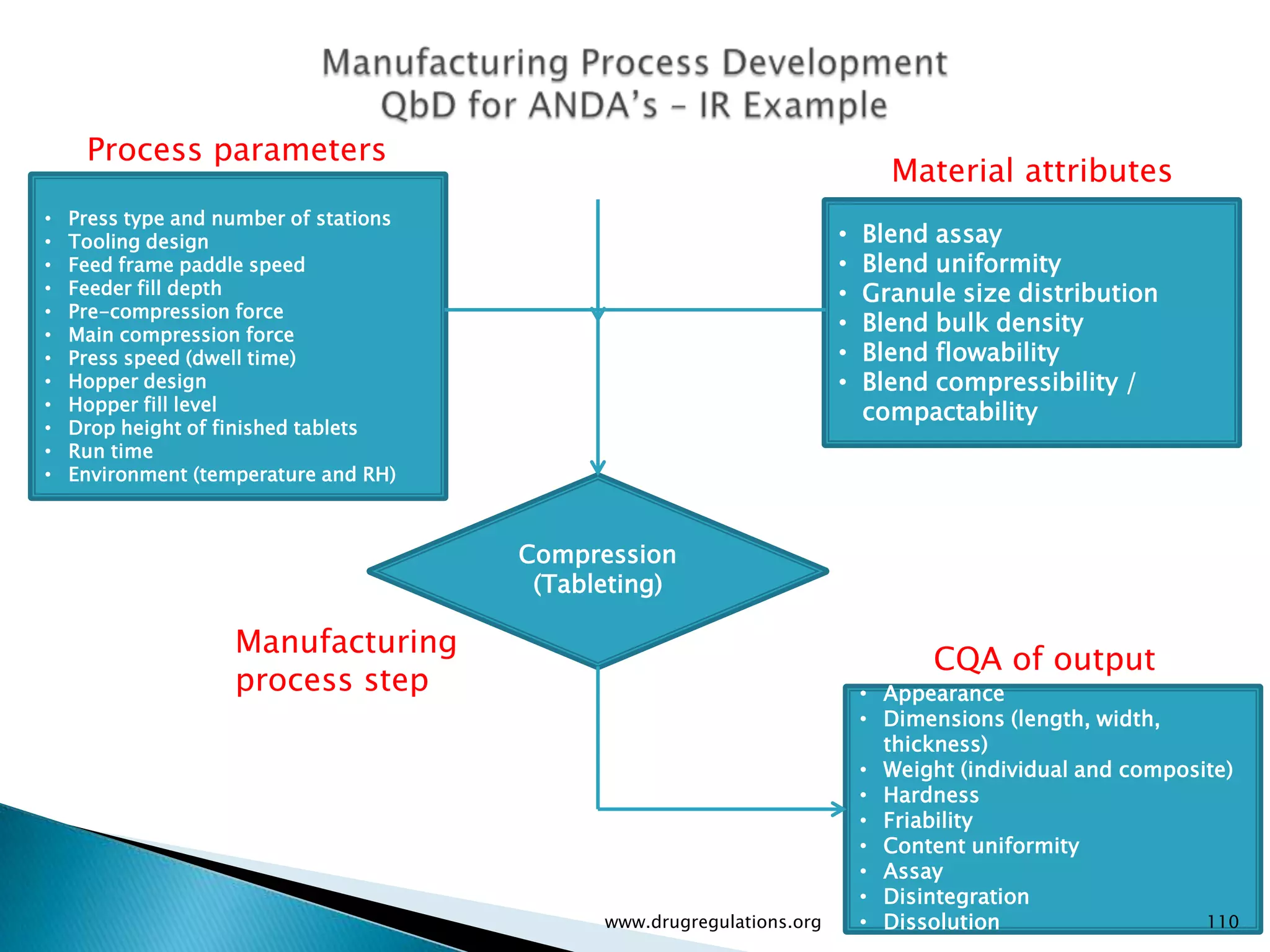
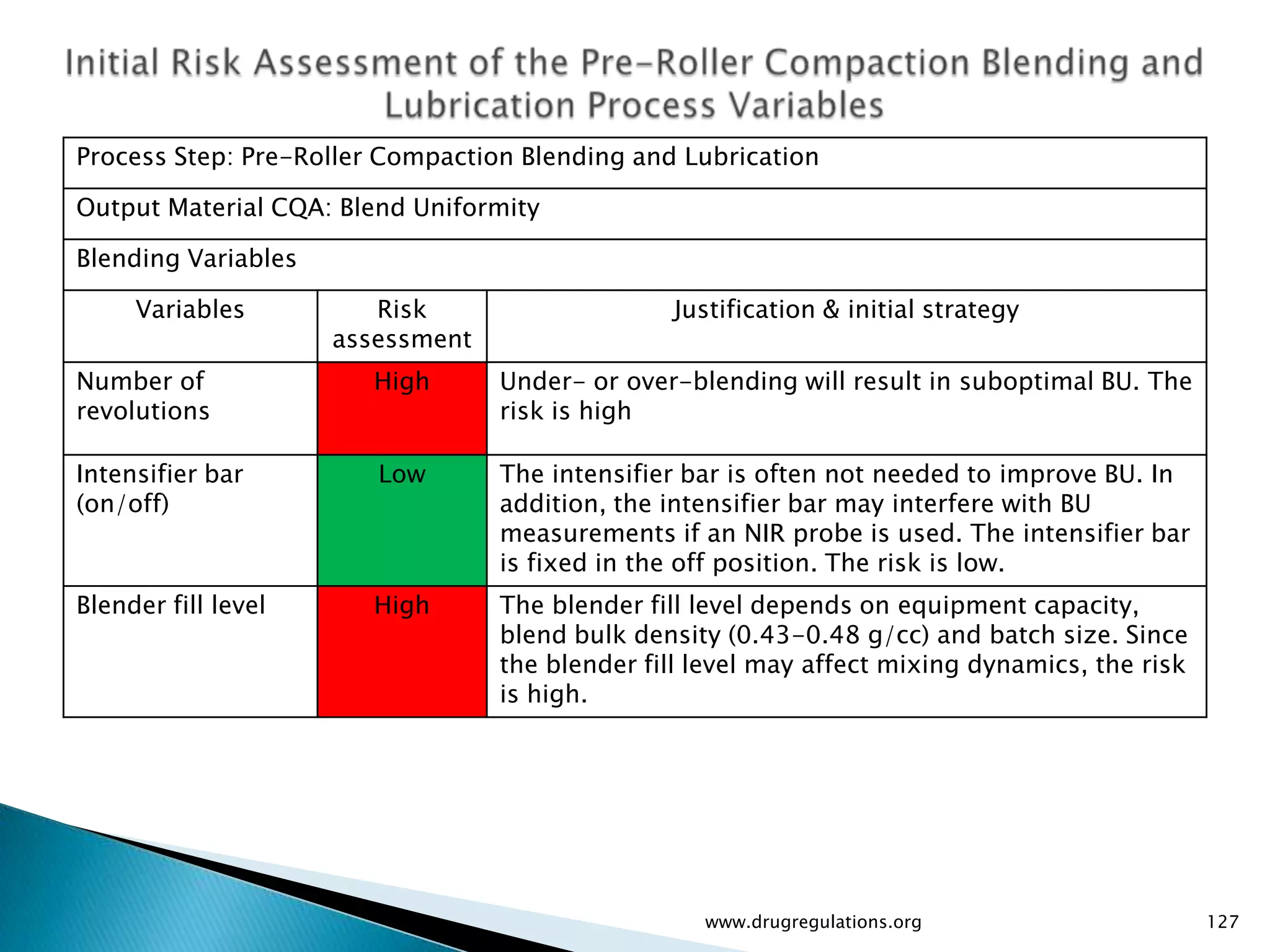
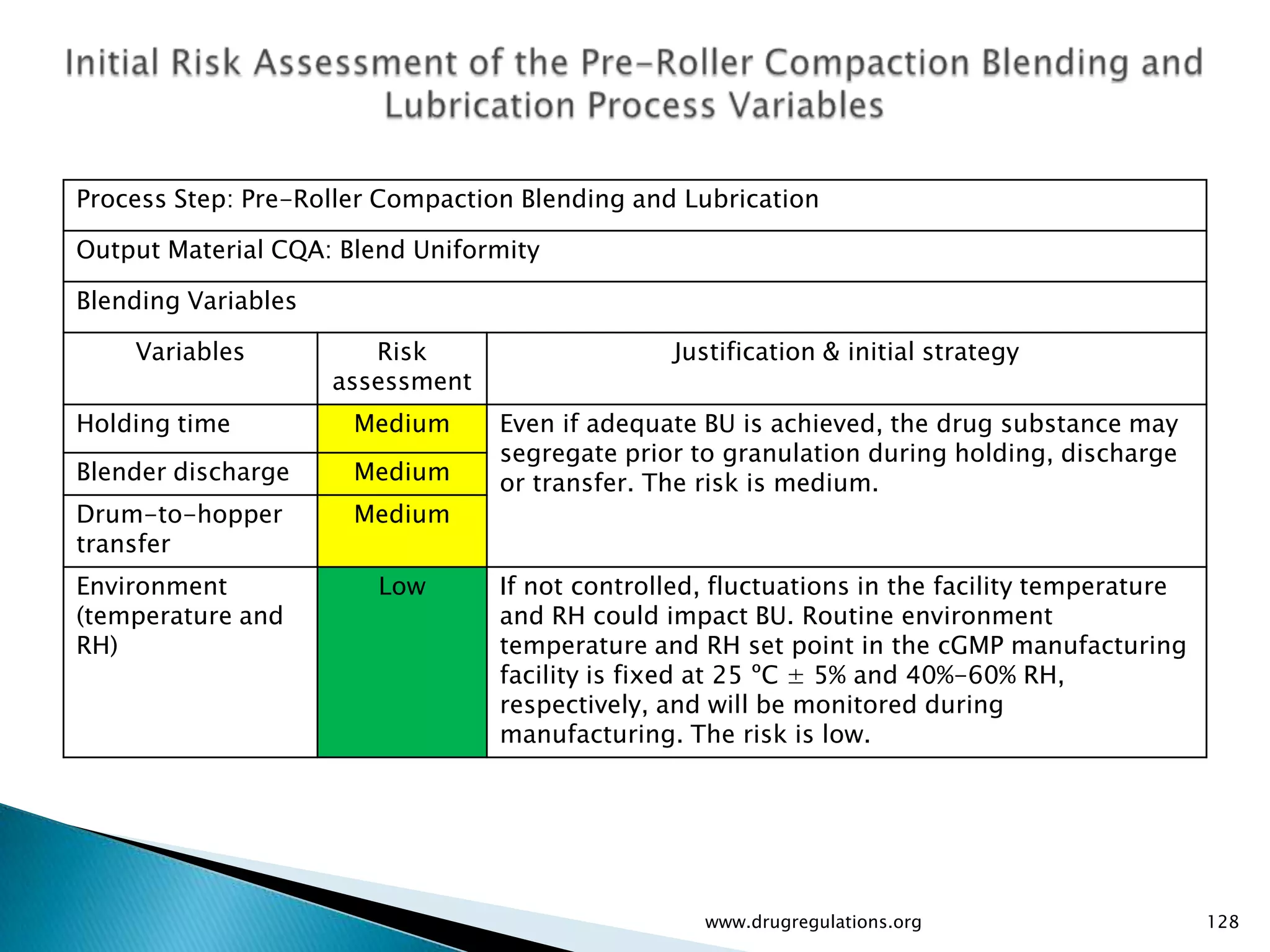
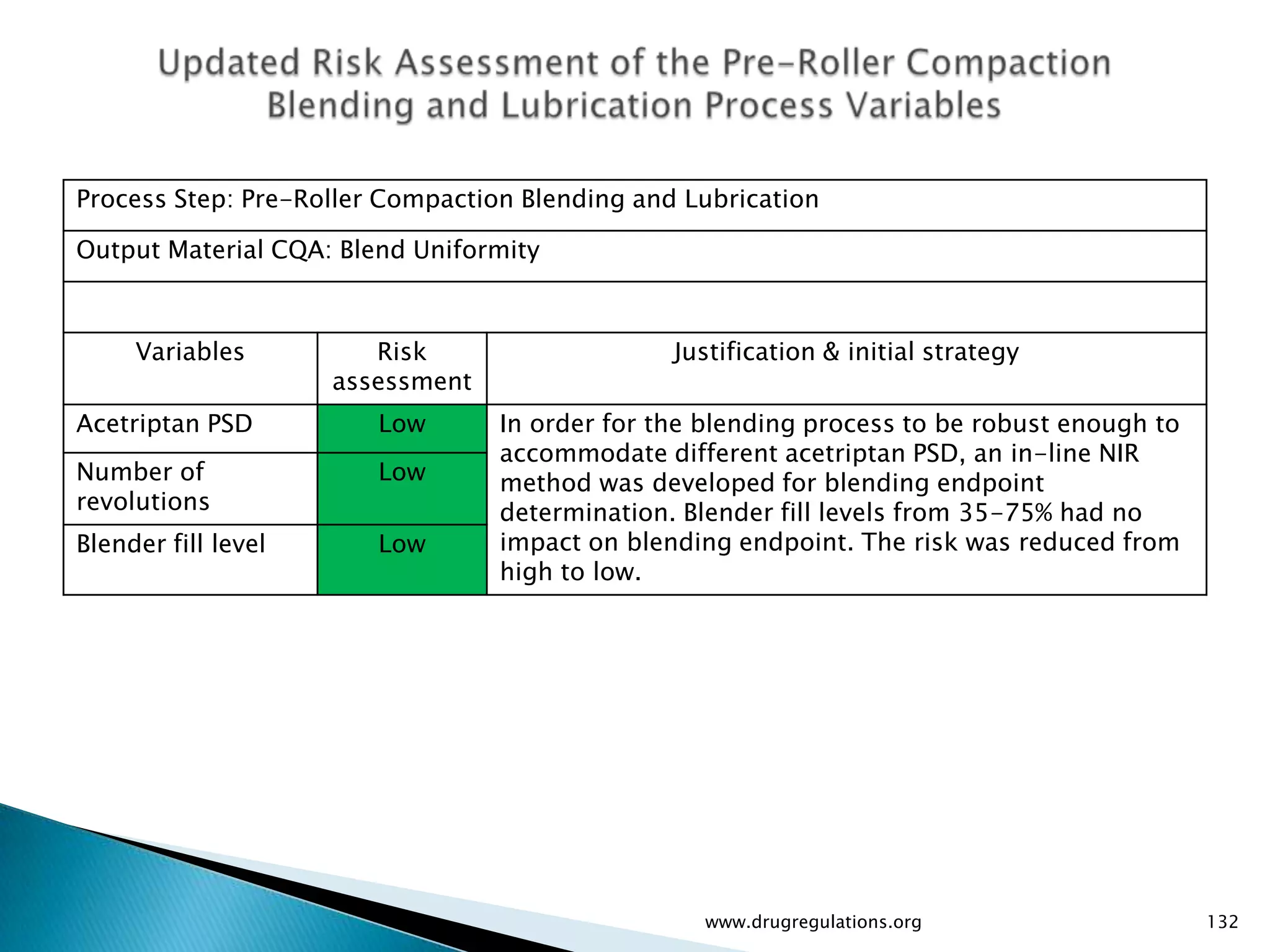
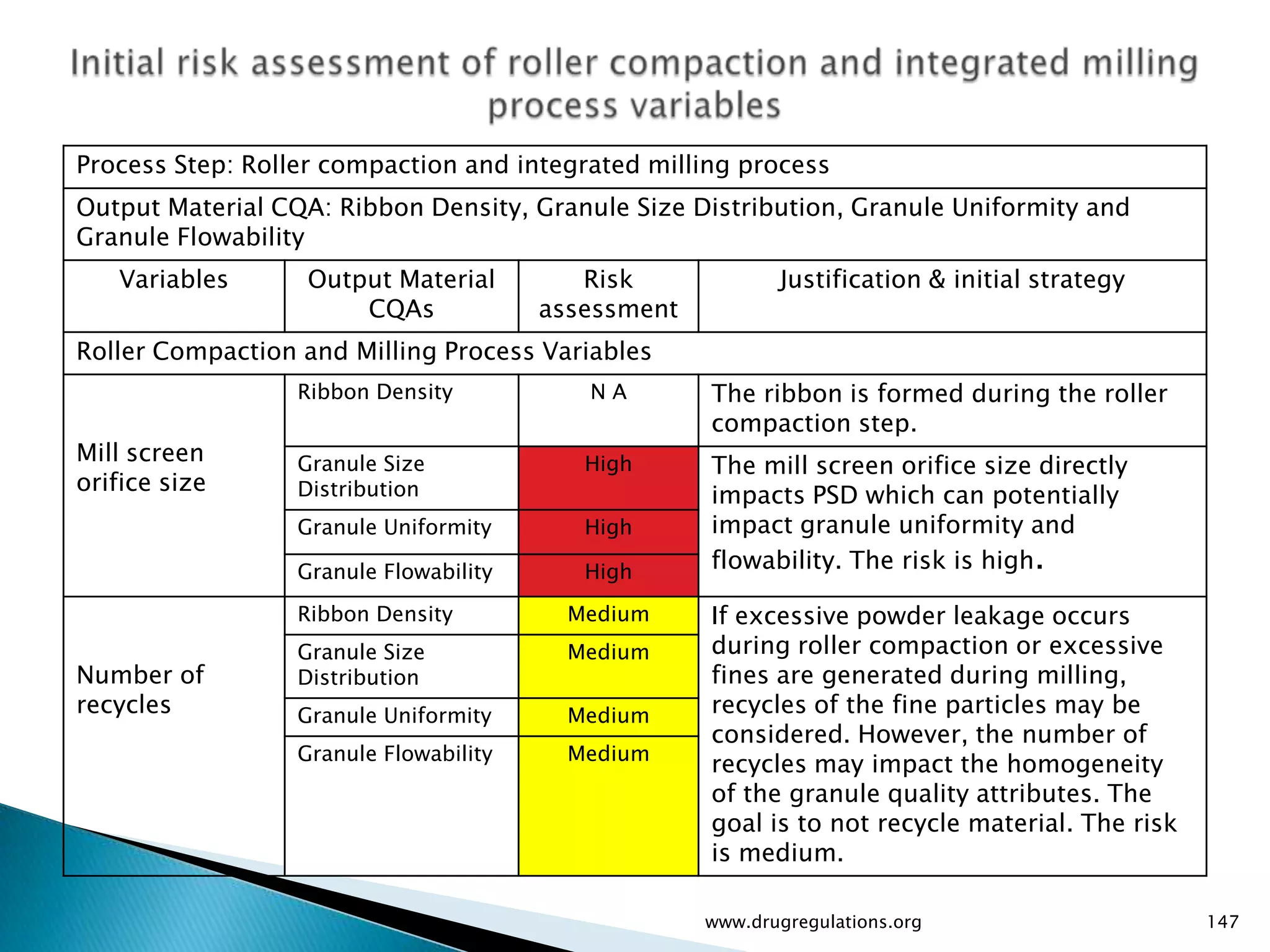
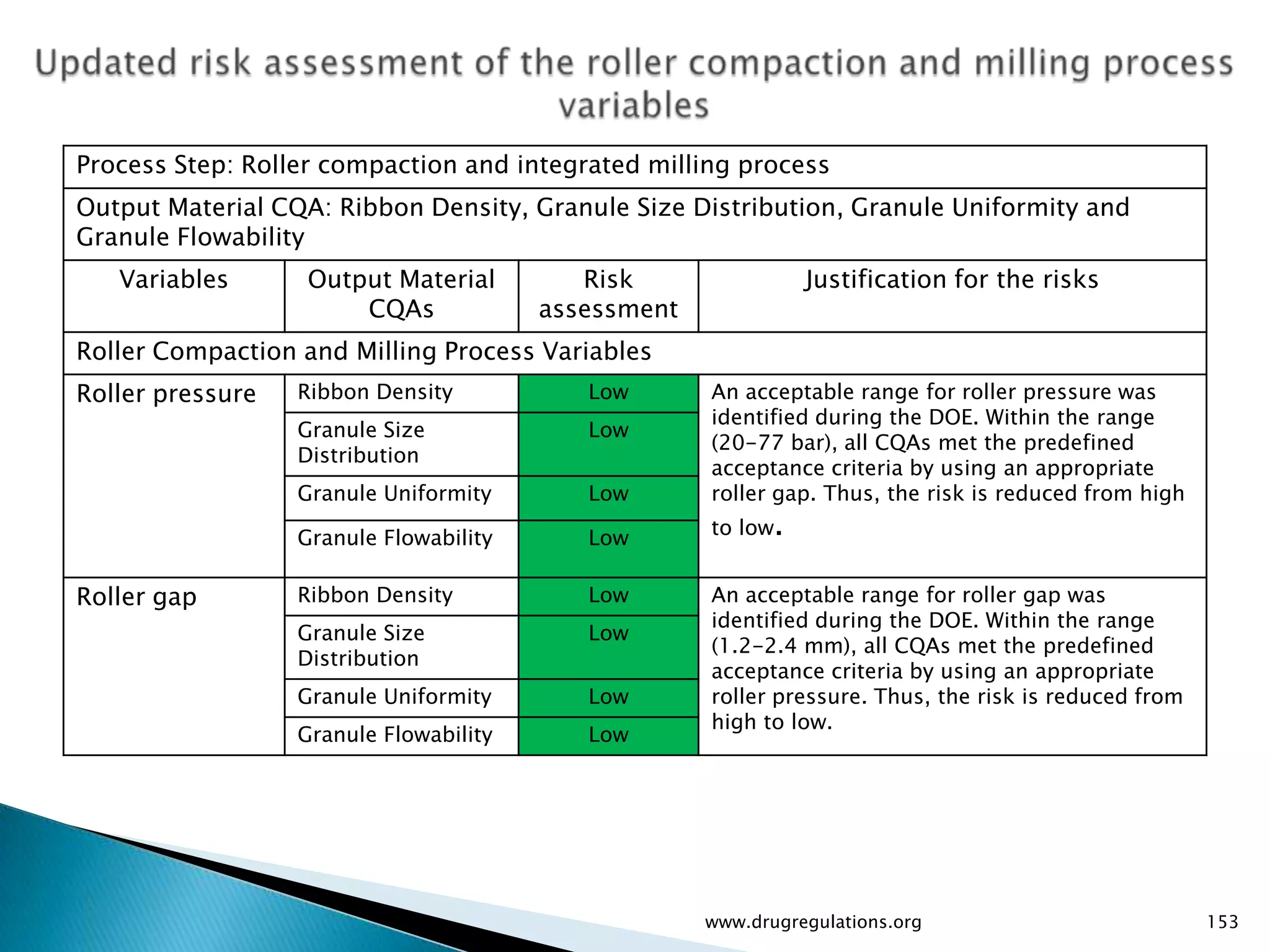
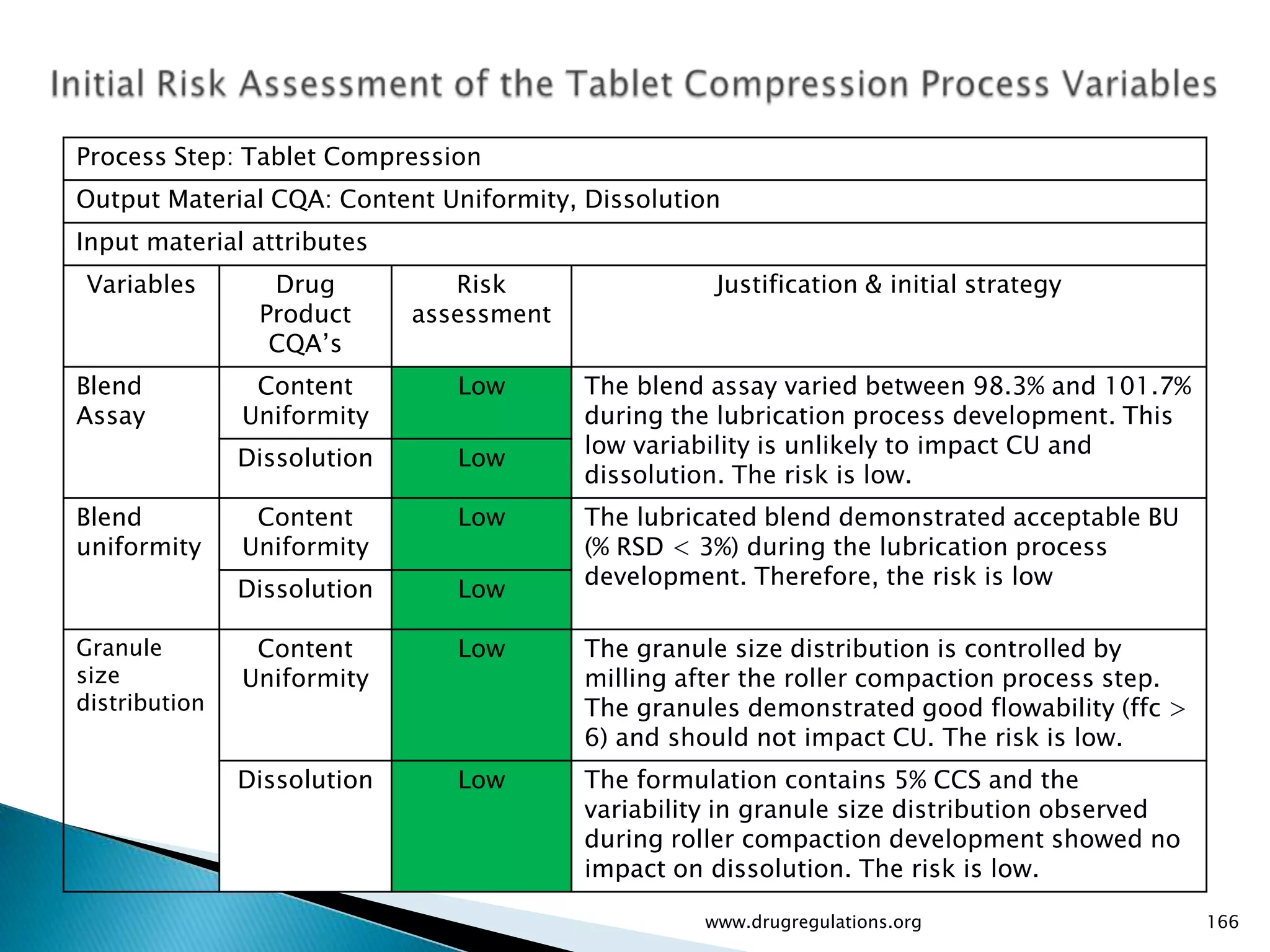
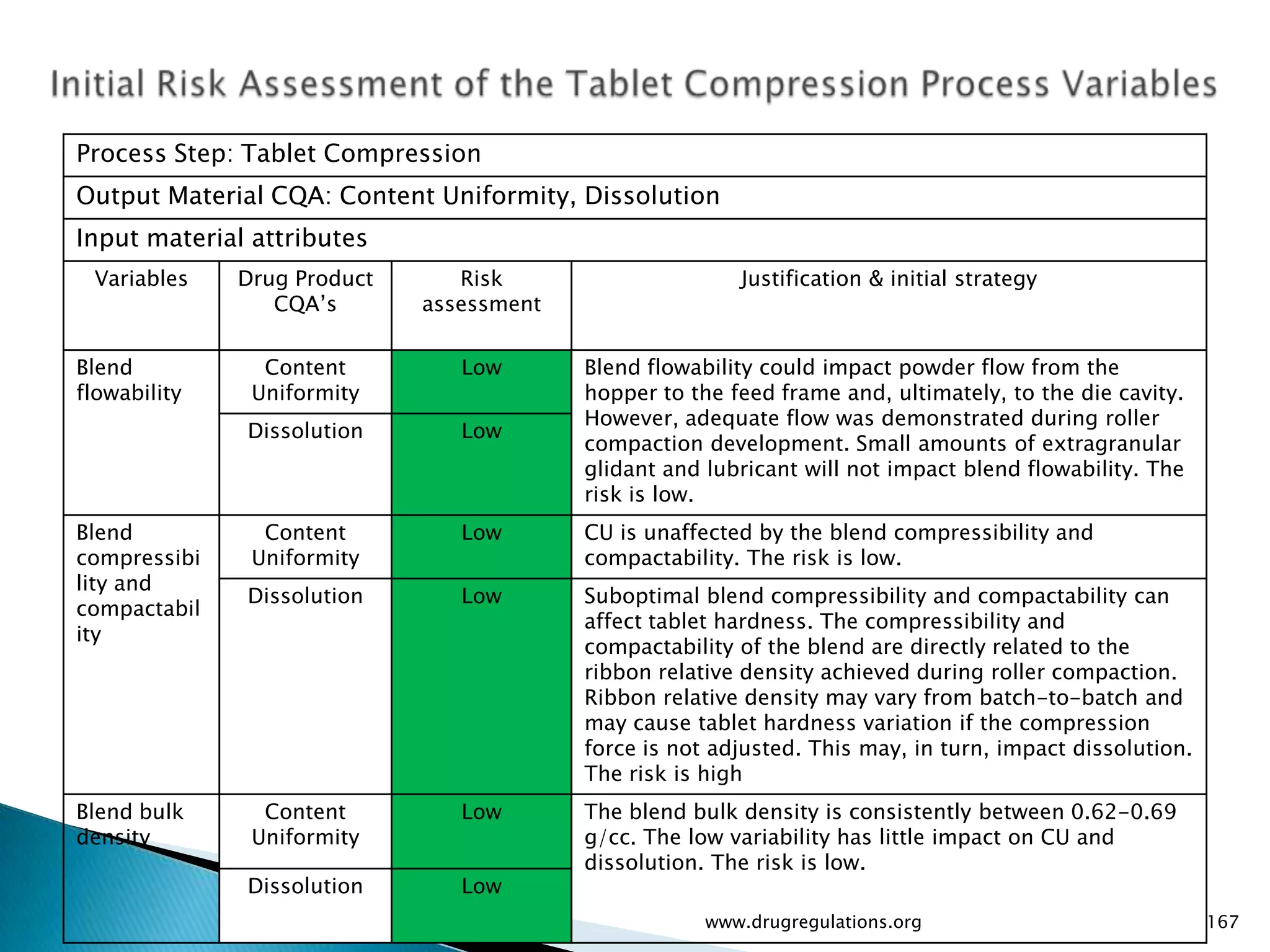
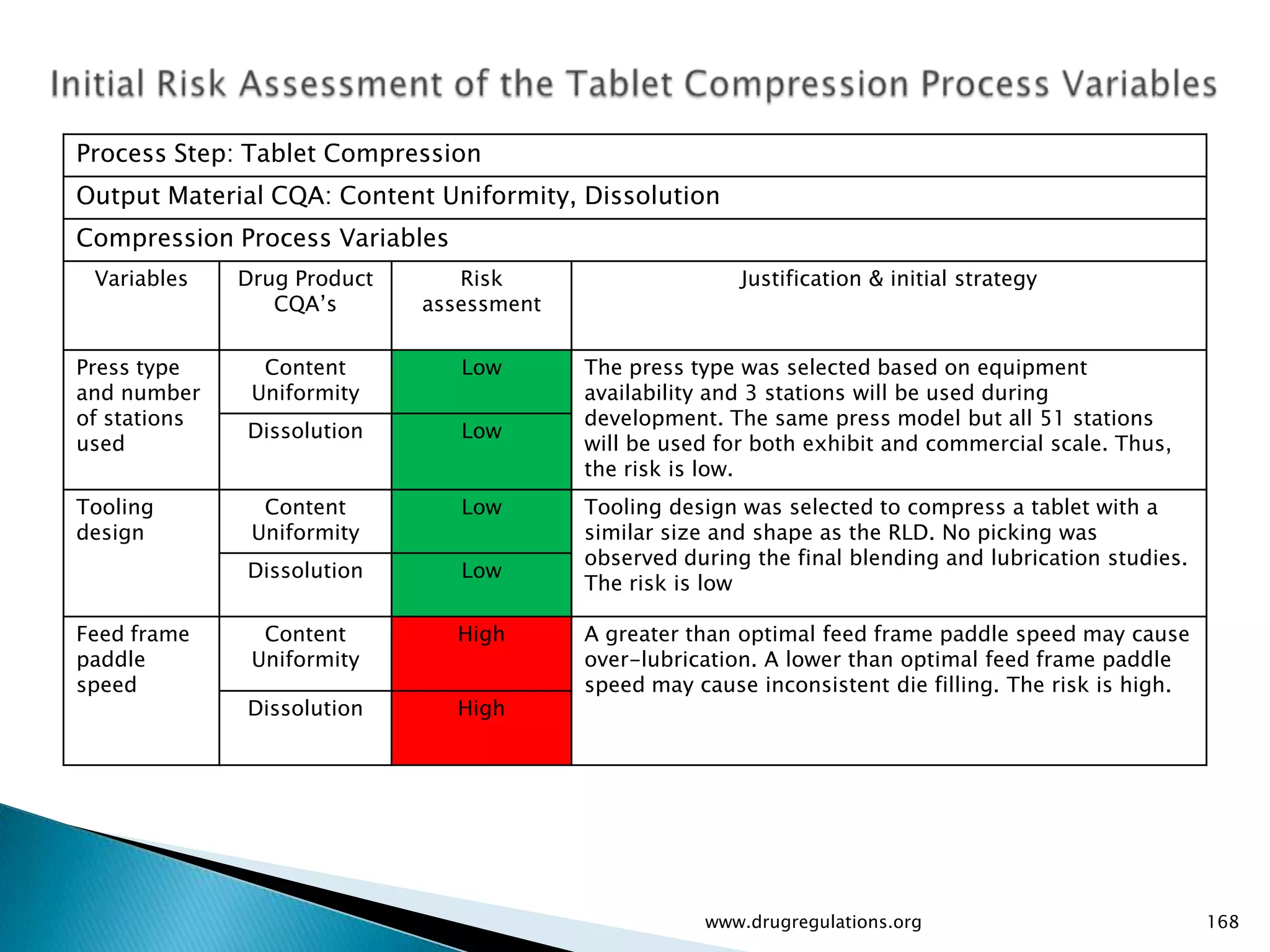
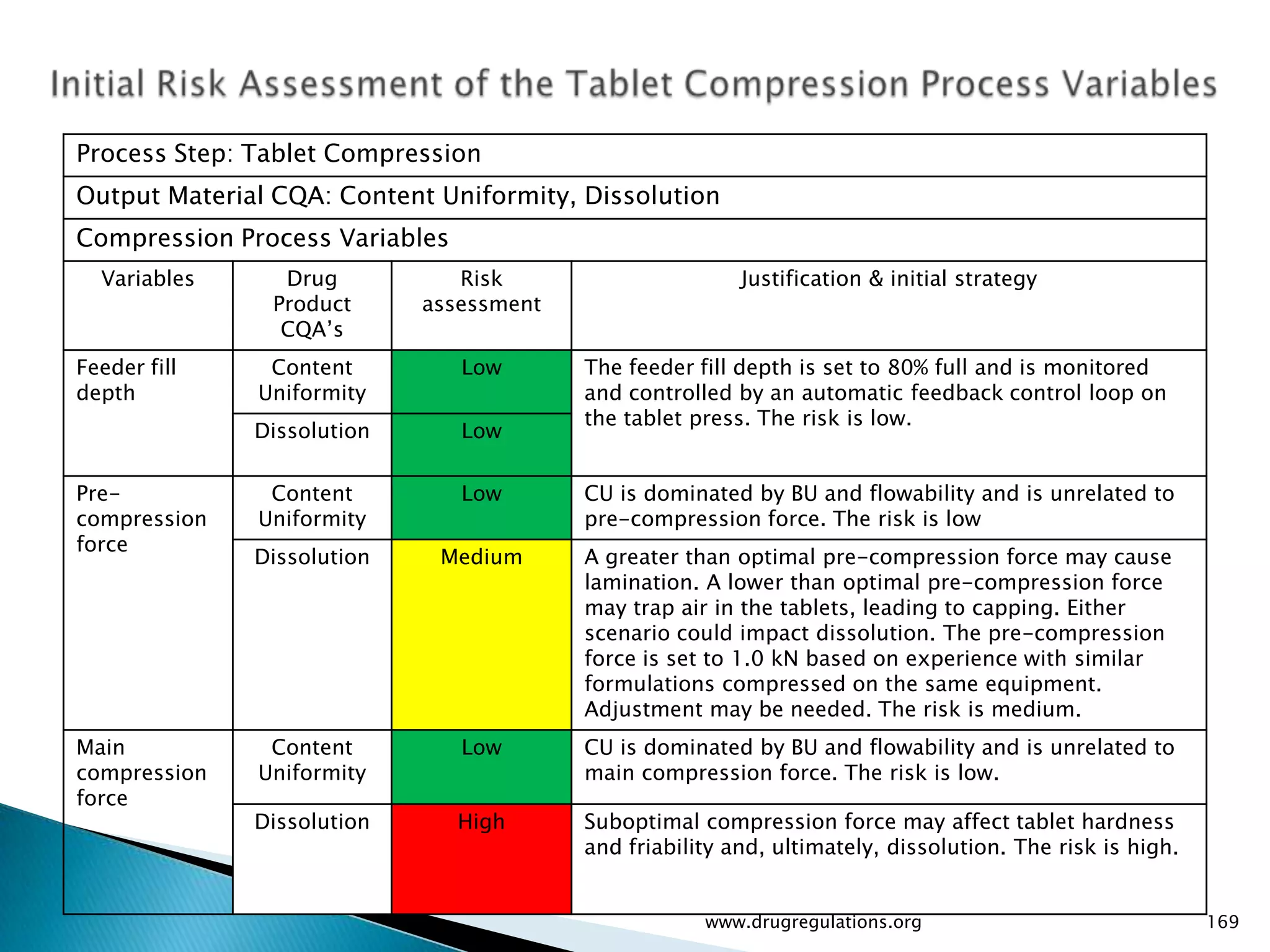
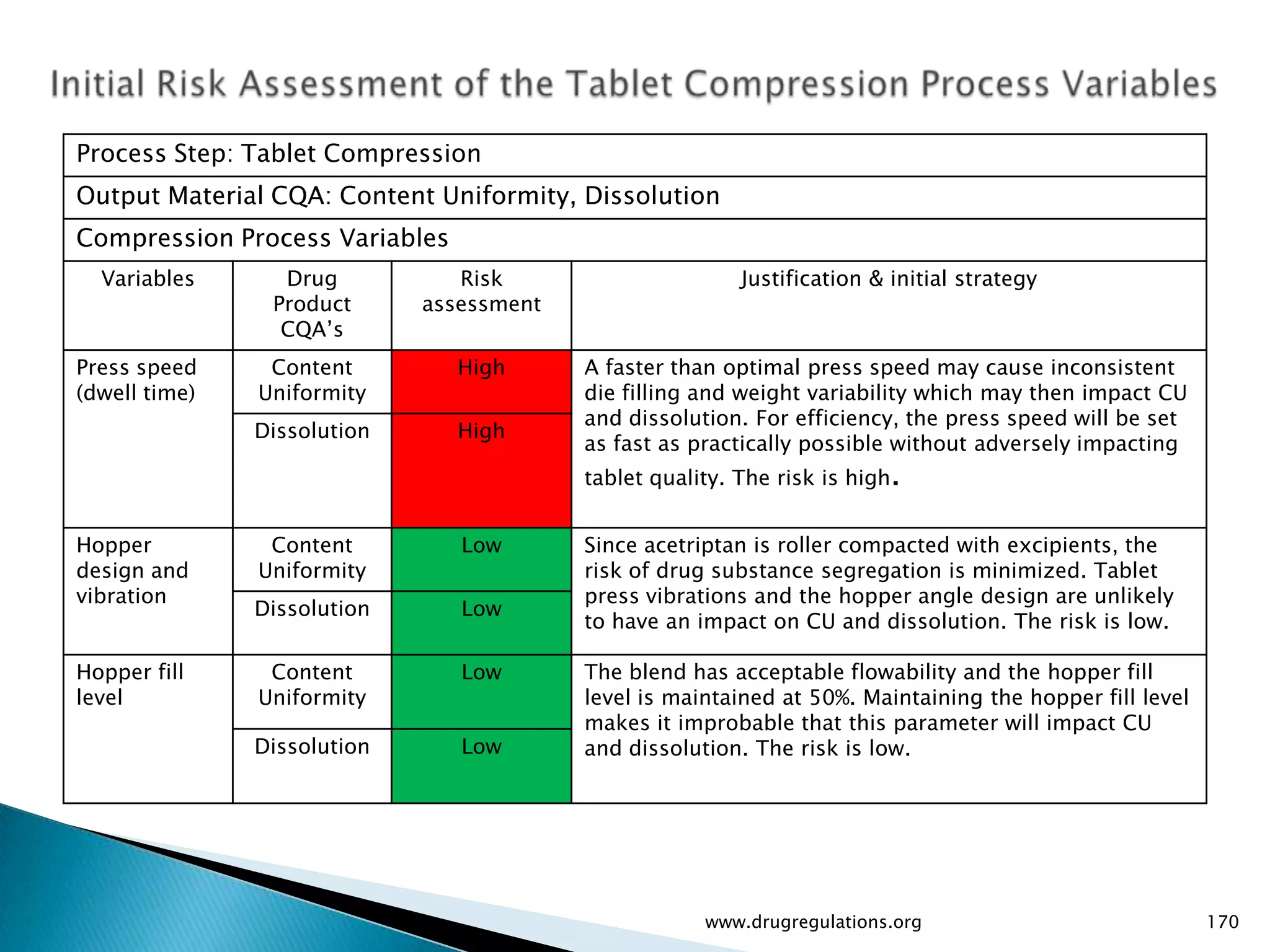
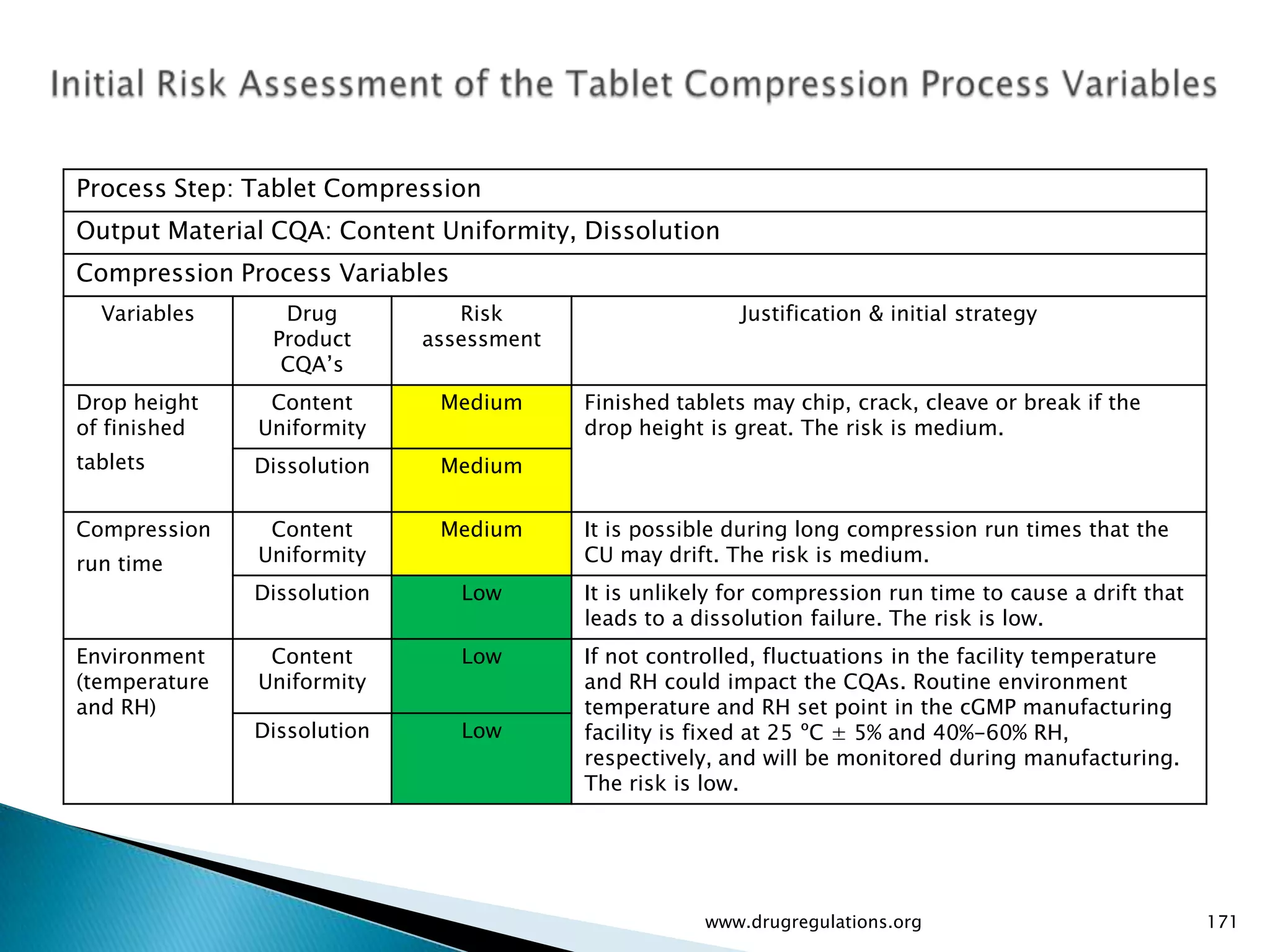
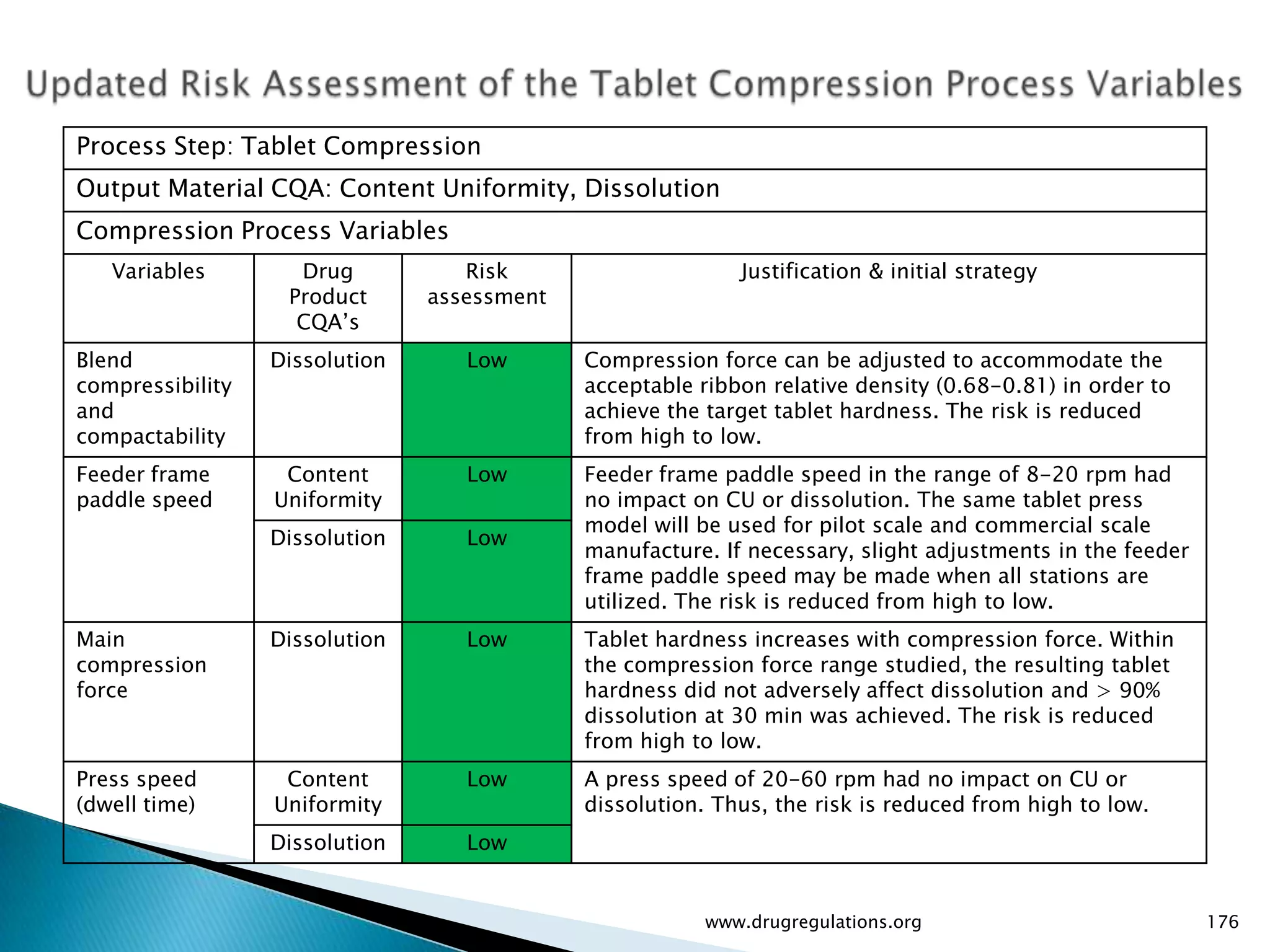
Summary of the tablet compression process variables affecting content uniformity and dissolution, highlighting risk management in manufacturing.
















































































![Where to Buy LinkedIn Accounts_ [12 Best Sites] (3).pdf](https://cdn.slidesharecdn.com/ss_thumbnails/wheretobuylinkedinaccounts12bestsites3-251124162550-95b6ddfa-thumbnail.jpg?width=640&height=640&fit=bounds)






